
One of our main interests in to provide the scientific community with tools that help with reproducible and open research. Ranging from complete pre-processing and downstream analysis pipelines for several types of genomic data, to interactive application to generate publication-ready figures.
We have developed a series of Snakemake pipelines that allow the user to process raw files (fastq) until a final html report, containing basic quality-control statistics, as well as downstream analyses (e.g., footprinting analysis for ATAC-seq data, peak enrichment por ATAC-seq and ChIP-seq).
All the instructions of how to use the pipelines can be found in the following GitHub repositories:
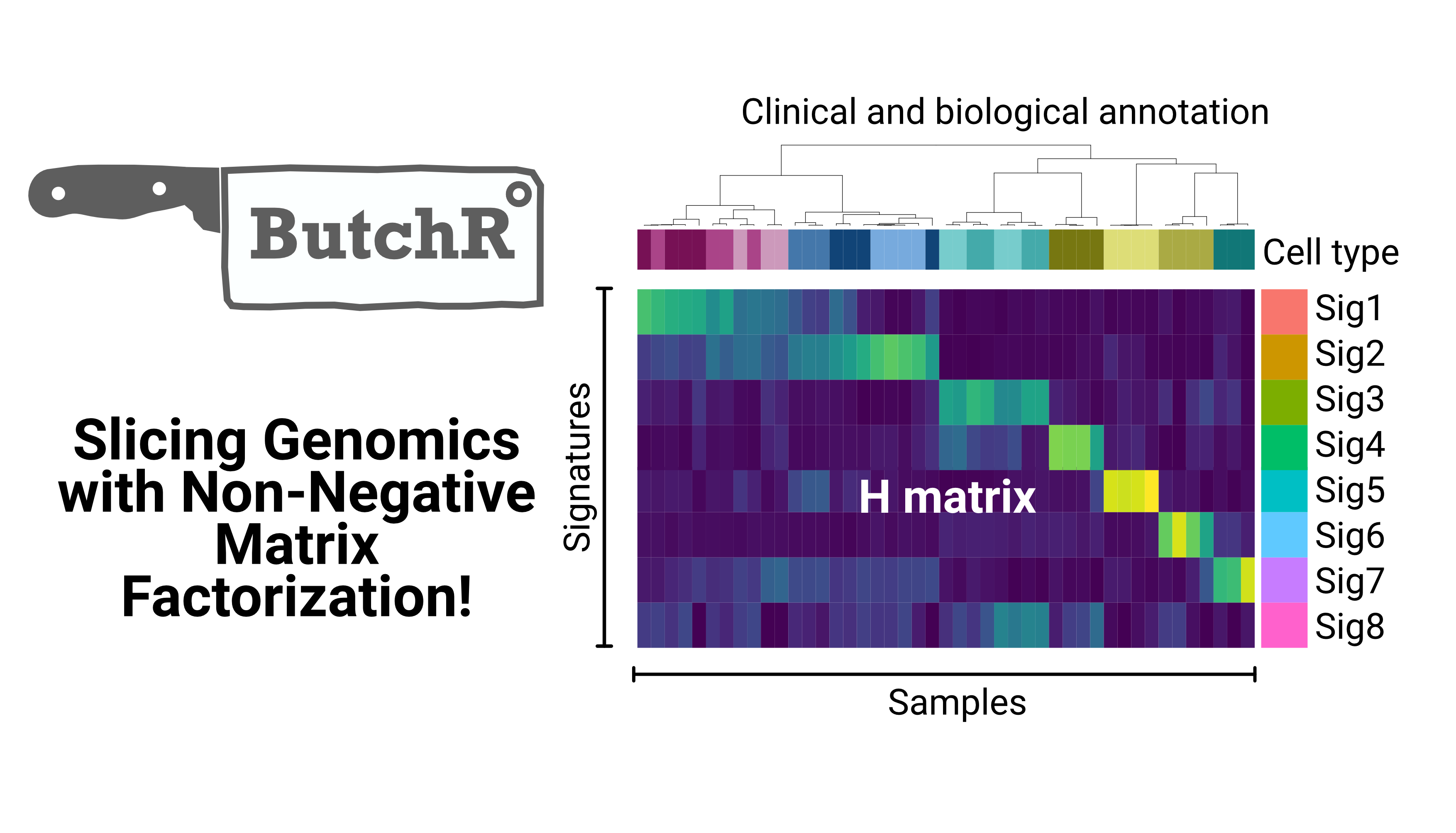
ShinyButchR is an interactive application to execute an NMF-based workflow from start to end. The results obtained with ShinyButchR can be imported into R to perform feature extraction and other downstream analyses.
The app is publicly available here, and we also provide a Docker image to allow its execution and deployment in local servers.
![]()
OntoVAE (Ontology guided VAE) is a VAE that can incorporate any biological ontology in its latent space and decoder, making the neurons interpretable. It can be used to compute pathway activities and to simulate perturbations. Our preprint on OntoVAE can be found here here.
The tool is available here. We also provide a vignette which explains step-by-step how to set up a custom OntoVAE workflow, with integrating an ontology, training a model on custom data, and carrying out perturbation experiments.
![]() The current SARS-CoV-2 pandemic has shown an impressive speed of spreading throughout the world. One of the main challenges is to understand how the viral sequence is evolving day by day. There are now multiple strains that are active at the same time, and keeping track of the population dynamics is not an easy task.
In order to address these challenges, we joined in the world effort to fight COVID-19 by developing MapMyCorona to display in an easy way, the sequence similarity and alterations between a query sequence and a central database of viral SARS-CoV-2 sequences on a world map.
The current SARS-CoV-2 pandemic has shown an impressive speed of spreading throughout the world. One of the main challenges is to understand how the viral sequence is evolving day by day. There are now multiple strains that are active at the same time, and keeping track of the population dynamics is not an easy task.
In order to address these challenges, we joined in the world effort to fight COVID-19 by developing MapMyCorona to display in an easy way, the sequence similarity and alterations between a query sequence and a central database of viral SARS-CoV-2 sequences on a world map.