Welcome to the Computational Regulatory Omics Lab (a.k.a. CROmLab) @ IPMB !
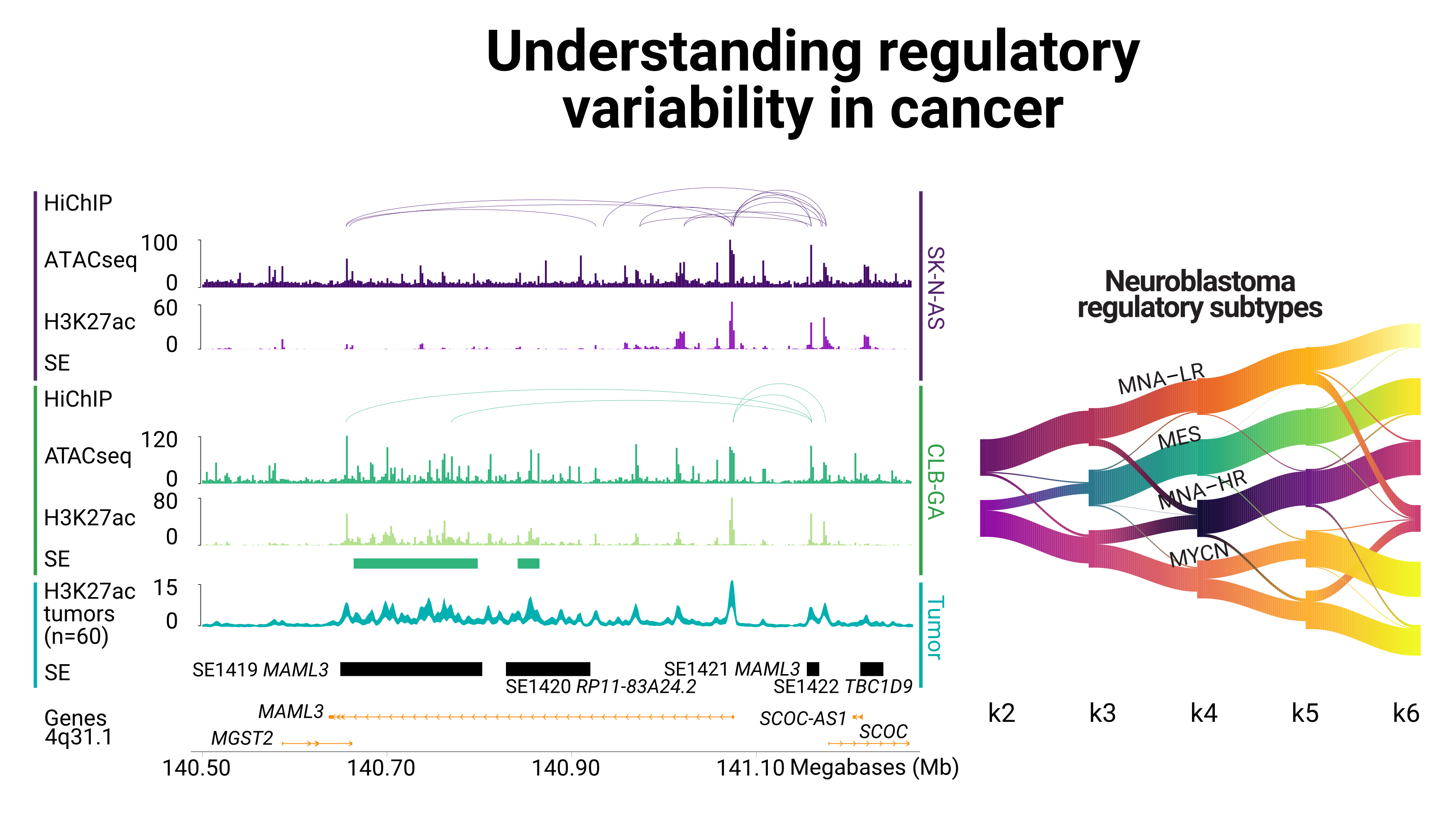
We are part of the Institute for Pharmacy and Molecular Biotechnology and BioQuant Center . We are working on understanding gene (de)regulation in disease, in particular cancer, mental disorders and infection.
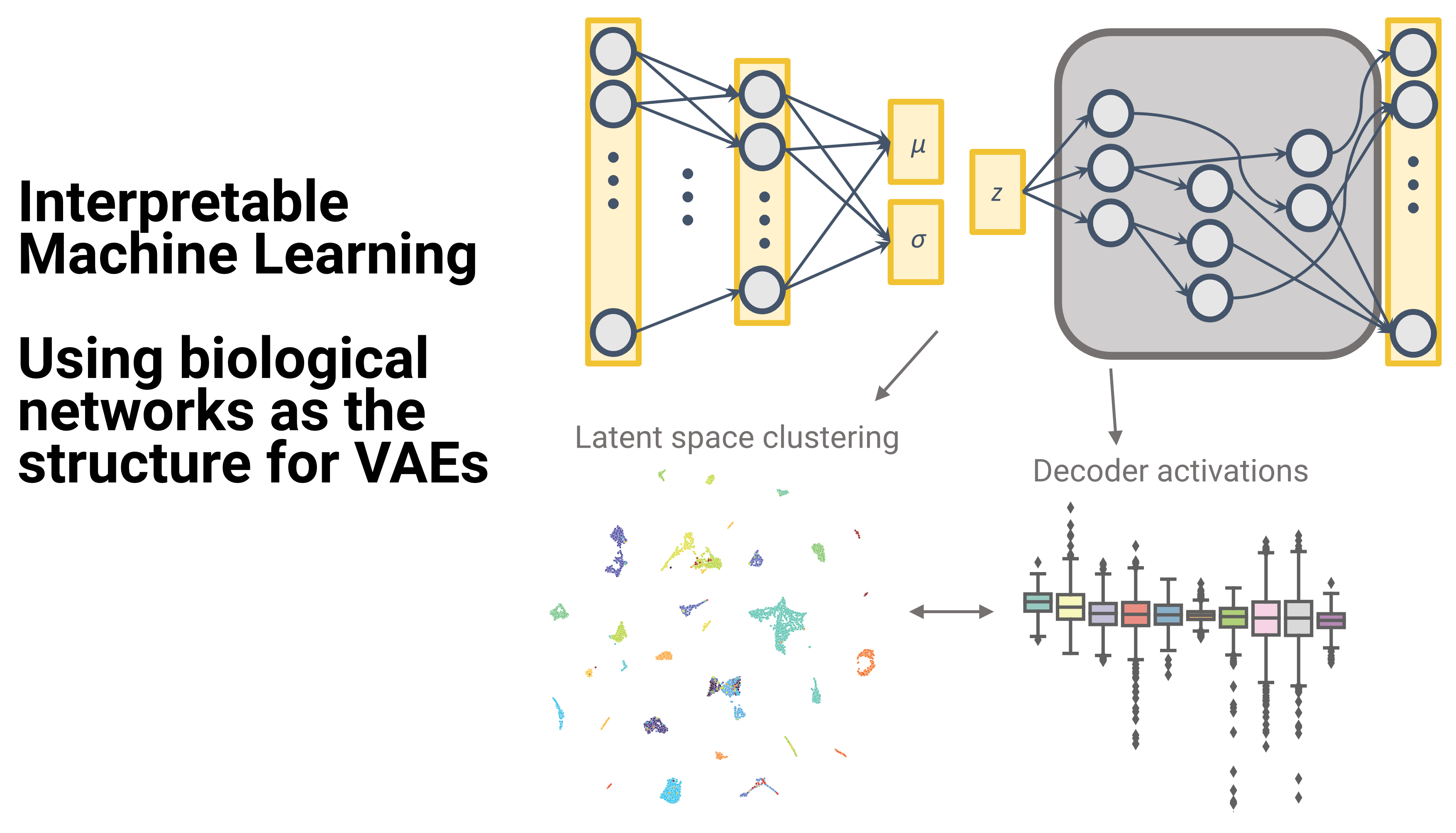
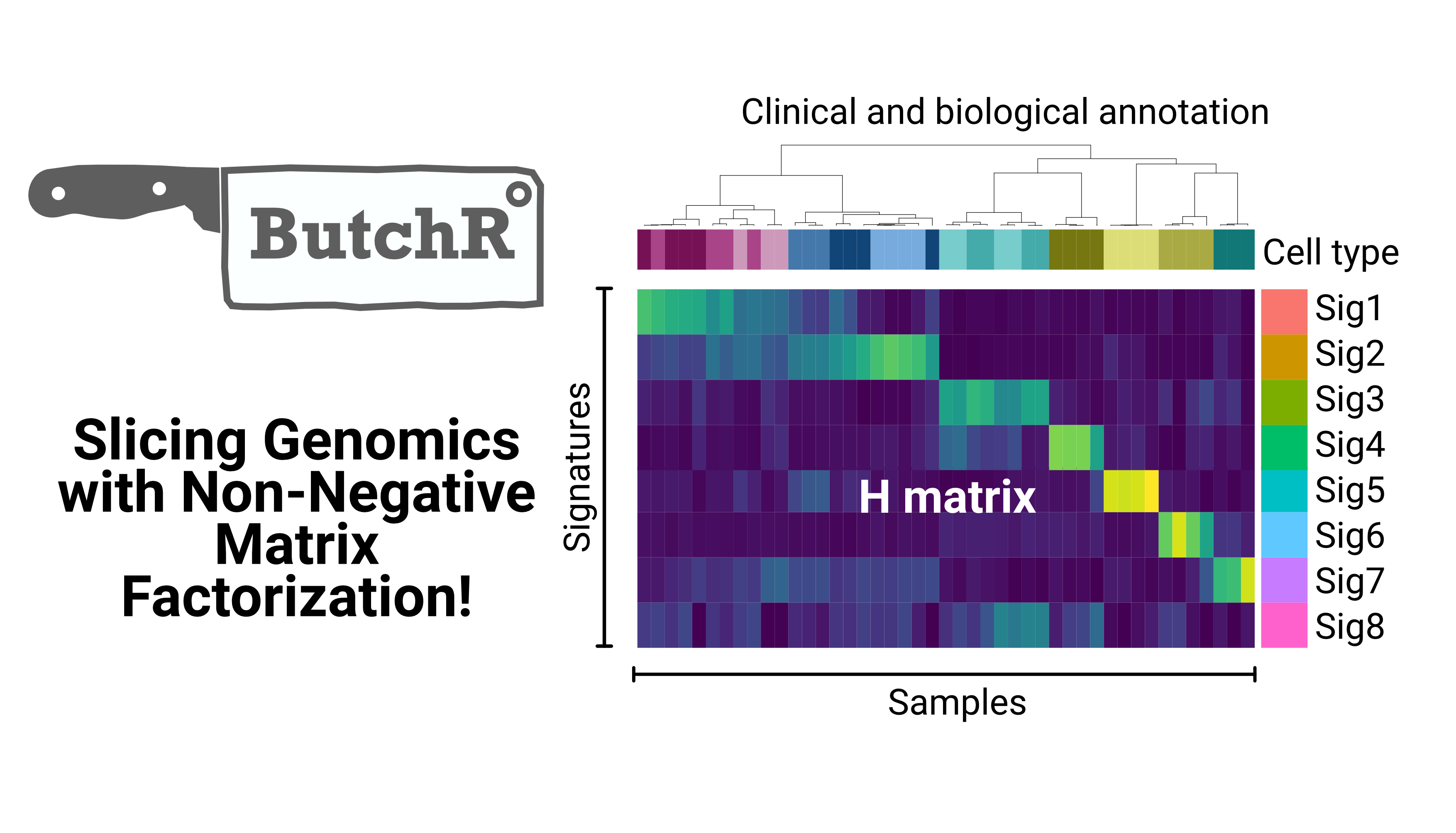
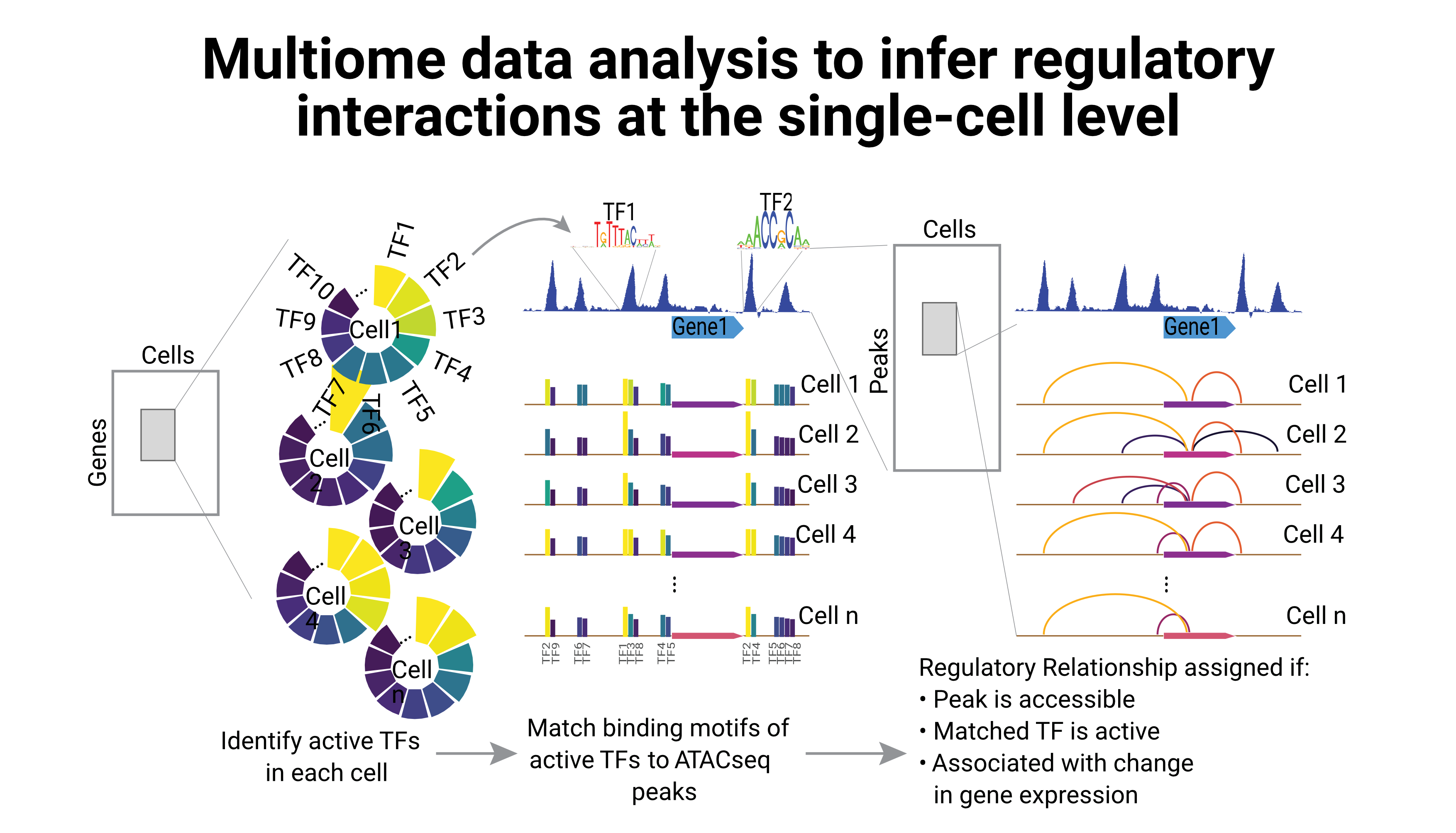
We develop novel methods to integrate large scale genomics datasets, using statistical and machine-learning approaches. We focus in particular on epigenomics data and single-cell approaches.
We are always interested in hosting bachelor/master/rotation students! Do not hesitate to contact us if you are interested in topics such as

For the 3rd time, we are organizing the DeepLife Workshop as part of the 4EU+ funded DeepLife project, with participants from Paris, Prag, Milan and Heidelberg. Check the website for more details!
May 2026Welcome Jan-Erik Bökenkamp, our new postdoc workin on a collaborative european project on multi-omics for papillary renal cell carcinoma!
July 2025Finally, our paper on a new hypermethalytion phenotype in glioblastoma came our in Genome Biology!
July 2025If you wonder if prediction methods for single-cell perturbation work, check out our new preprint on BioRxiv!
13. May 2025Thien has successfully defended his Master thesis today! Happy to keep him as a PhD student in the group!
10. September 2024Congratulations Youcheng for defending your PhD thesis today! Very happy to have you with us for some more time!
12. June 2024We are hosting the DeepLife Hackathon as part of the 4EU+ funded DeepLife project. Check the website for more details!
1. May 2024Welcome to Jean, our new PhD student working on the Carl-Zeiss foundation funded project on tumor cell plasticity (AI-CARE)!
2. April 2024Youcheng's paper on networks of comorbidity signatures has been published in Discover Mental Health! This is part of the COMMITMENT project on comorbidities in schizophrenia. Congratulations Youcheng!