The Computational Regulatory Omics Labt at IPMB and BioQuant is working on understanding gene (de)regulation in disease, in particular cancer, mental disorders and infection. We develop novel methods to integrate large scale genomics datasets, using statistical and machine-learning approaches. We focus in particular on epigenomics data and single-cell approaches.
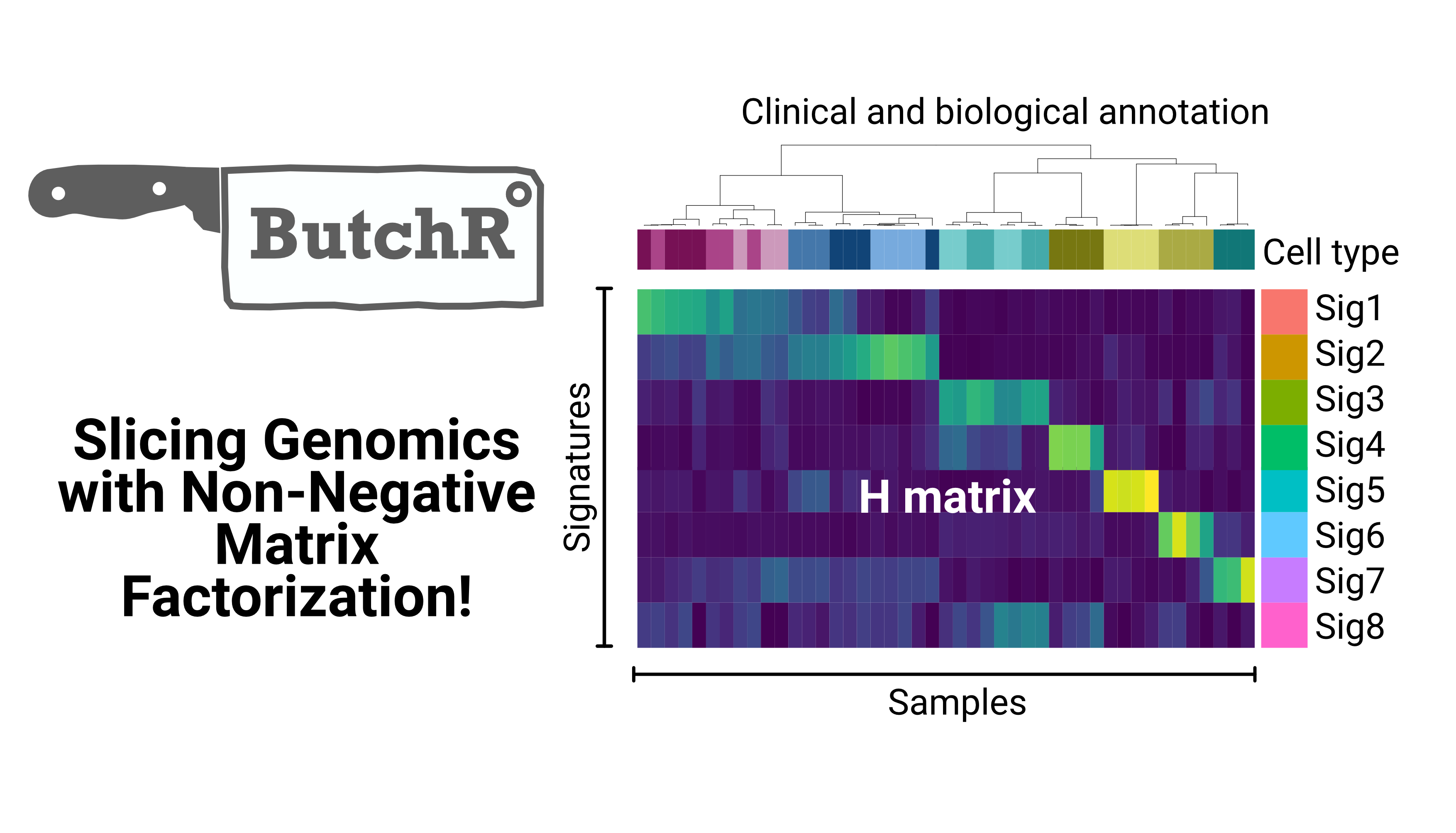
We developed ButchR, a new toolkit to infer signatures and extract relevant features associated with genotypes and phenotypes using non-negative matrix factorization (NMF). This tool can be used to extract a lower dimensional representation from multi-omics data layers in bulk and single-cell genomics.
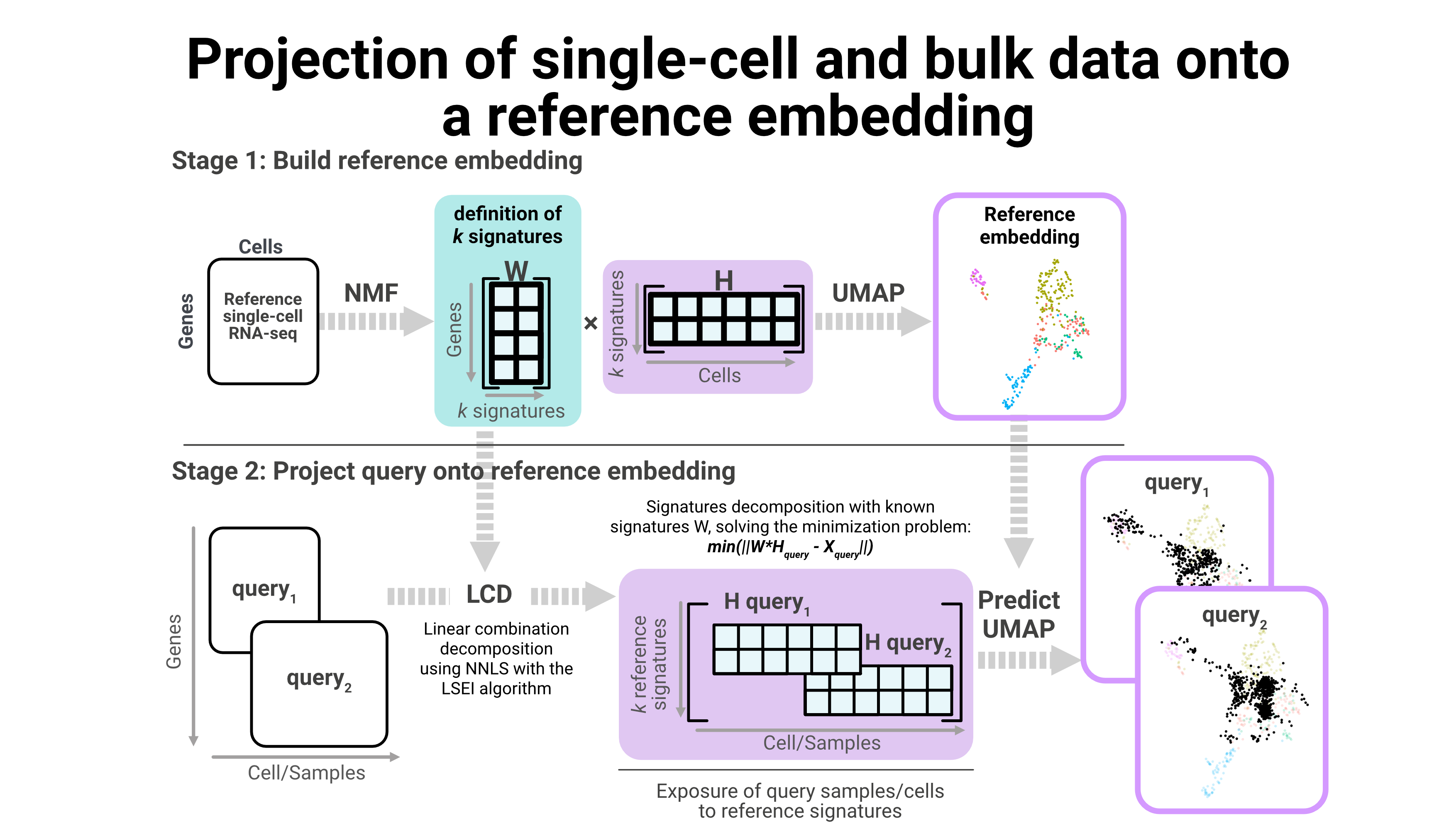 We are also developing new strategies to combine single-cell and bulk data, like projecting samples and cells into a reference embedding. Using this approach we have been able to understand the possible cell of origin for complex tumor identities.
We are also developing new strategies to combine single-cell and bulk data, like projecting samples and cells into a reference embedding. Using this approach we have been able to understand the possible cell of origin for complex tumor identities.
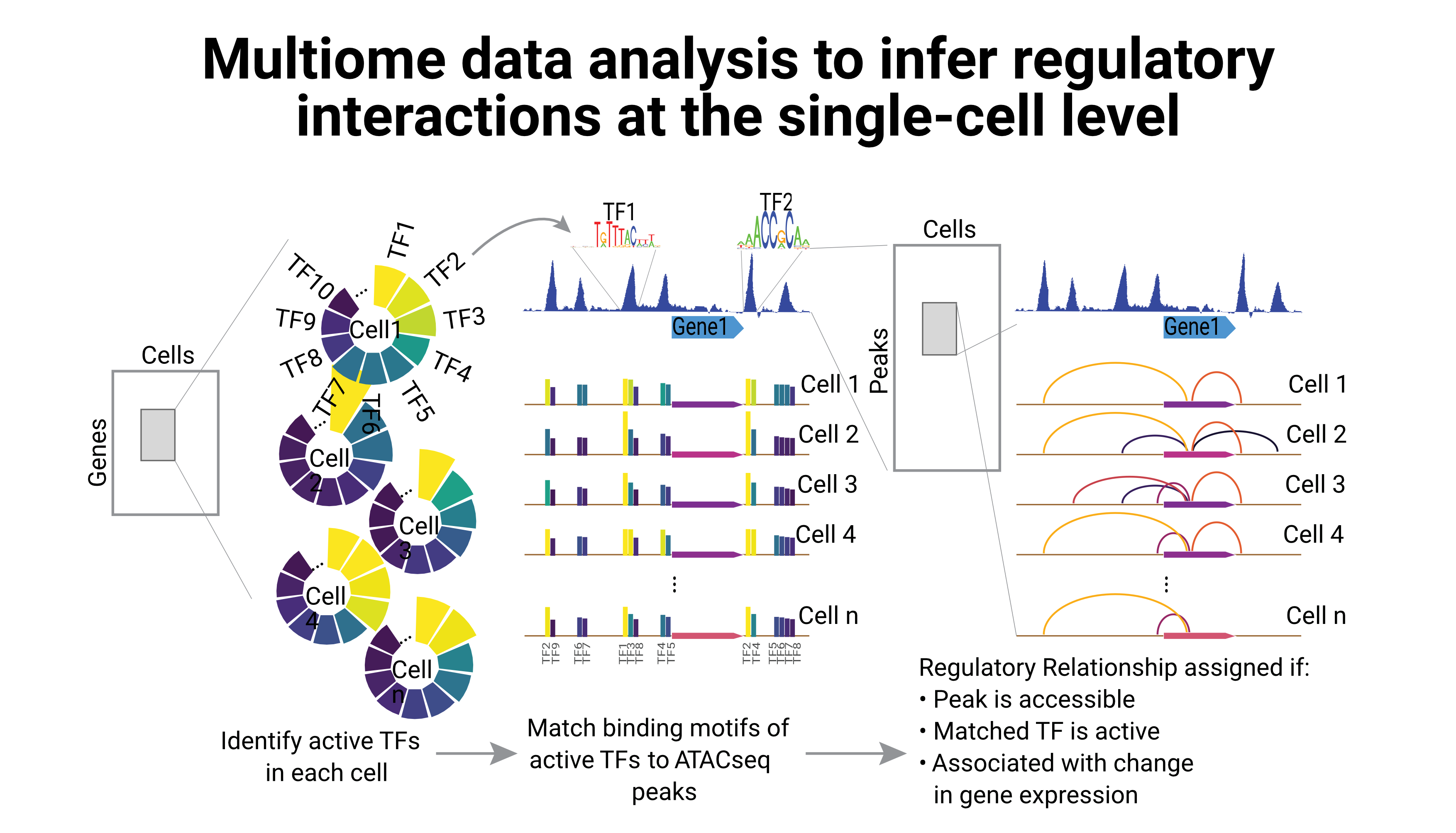 Regulation of gene expression is tightly coupled with the epigenetic landscape of the cell; but until recently, it was not possible to study the direct relationship between the transcriptome and the chromatin accessibility at the single-cell level. However, several multiome technologies have overcome this limitation, providing both layers of information within the same single cell.
Regulation of gene expression is tightly coupled with the epigenetic landscape of the cell; but until recently, it was not possible to study the direct relationship between the transcriptome and the chromatin accessibility at the single-cell level. However, several multiome technologies have overcome this limitation, providing both layers of information within the same single cell.
One of our main research interests is to use such data to infer the regulatory interactions between one gene and its cis-regulatory elements. We have used the inferred regulatory landscape on single-cells, to identify regulatory differences between different cell types.
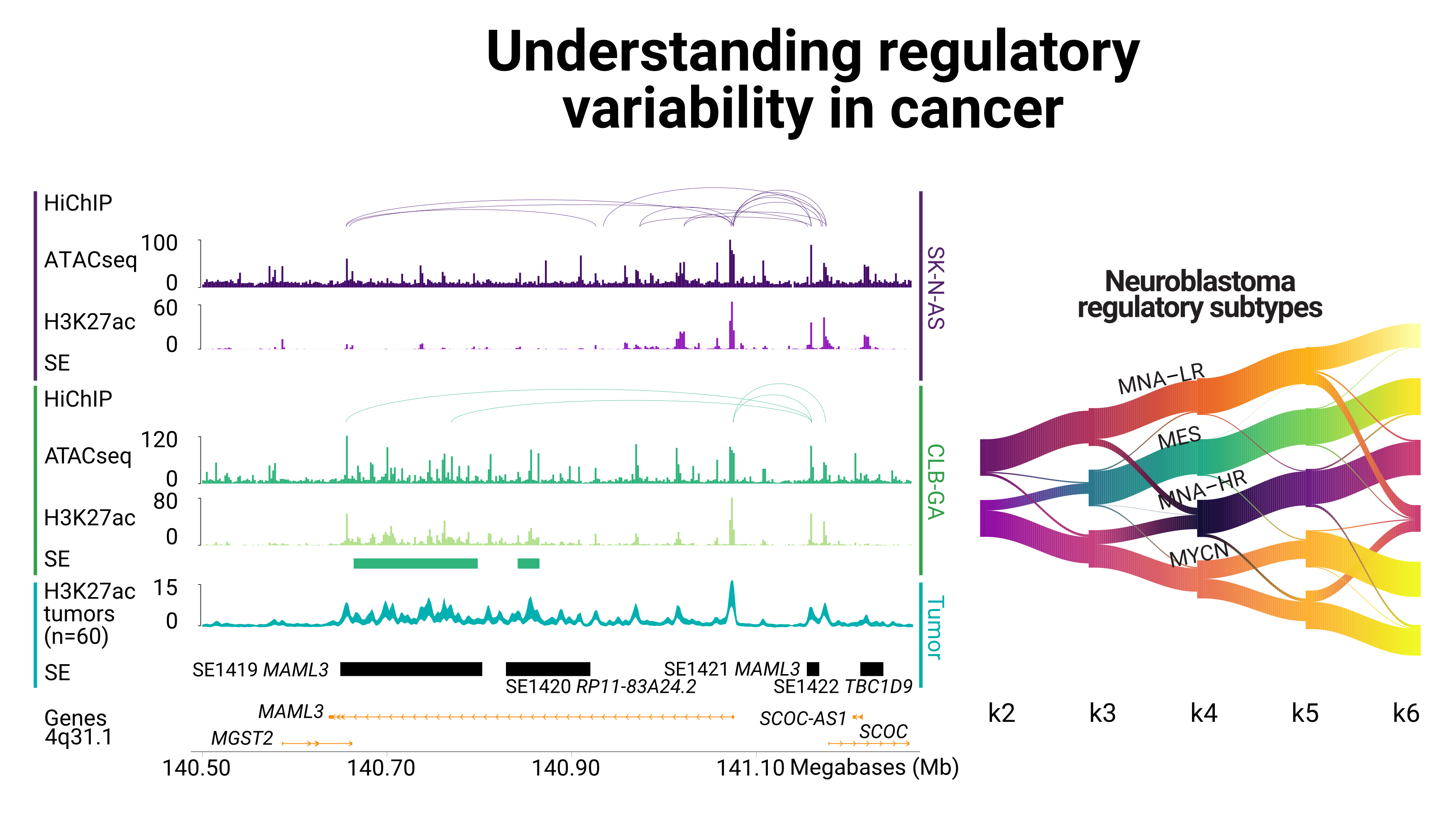 Neuroblastoma subtypes appear to be strongly defined through epigenomic profiles. We integrate ChIP-seq data on histone modification to extract specific signatures for NB subtypes from tumors and cell lines.
Neuroblastoma subtypes appear to be strongly defined through epigenomic profiles. We integrate ChIP-seq data on histone modification to extract specific signatures for NB subtypes from tumors and cell lines.
This work is done in close collaboration with the Division of Neuroblastoma Genomics at the DKFZ
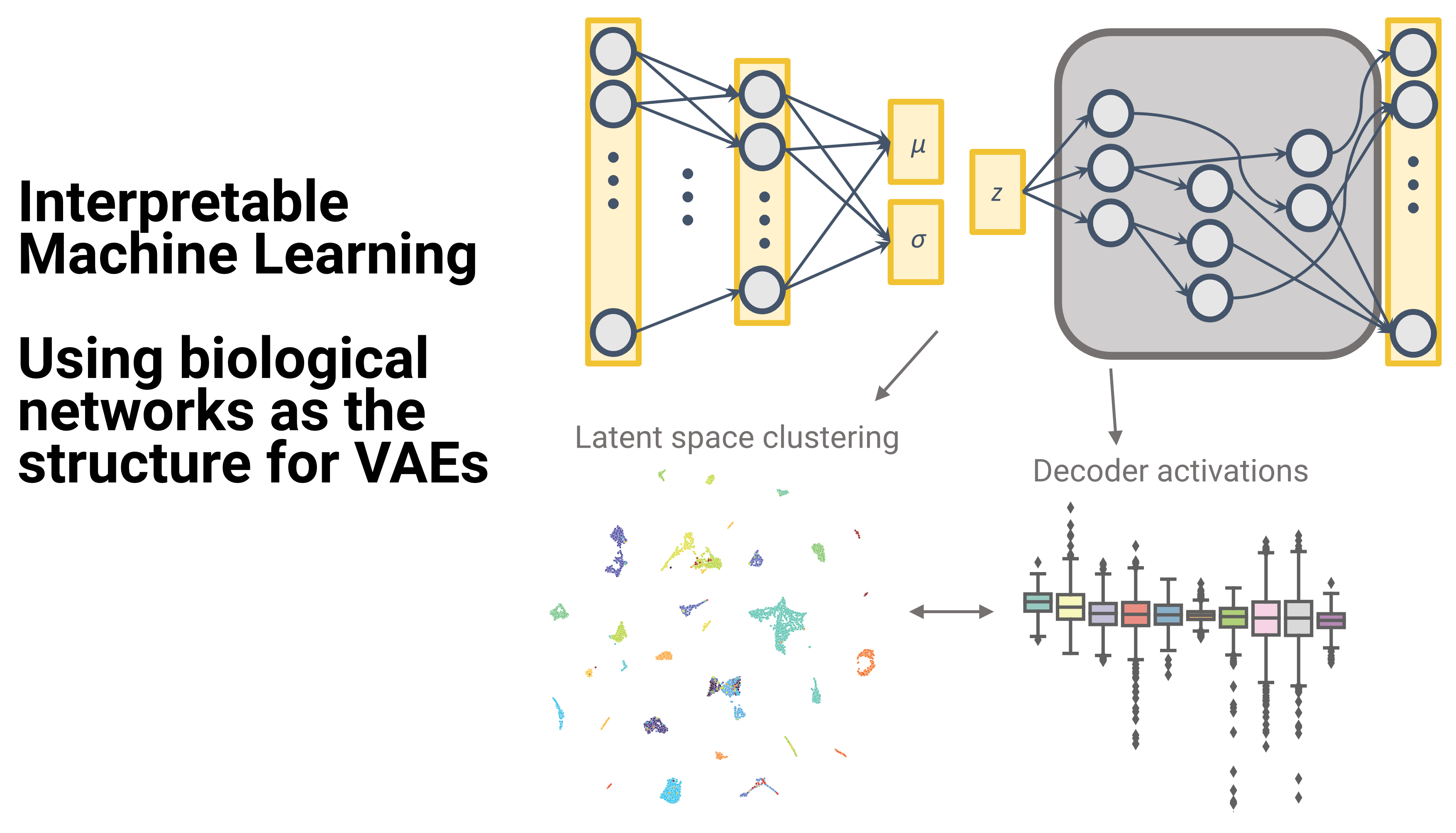
We are building Variational Autoencoders (VAEs) using biological networks as the structure for their decoder. Looking at the decoder activations we can determine which nodes in the nerwork are more relevant for a determined set of samples.