[BC]2 Tutorial
Defining genomic signatures with Non-Negative Matrix Factorization
Carl Herrmann & Andres Quintero
13 September 2021ButchR
ButchR is an R package providing functions to perform
non-negative matrix factorization and postprocessing using TensorFlow.
https://github.com/wurst-theke/ButchR
Citation
Andres Quintero, Daniel Hübschmann, Nils Kurzawa, Sebastian Steinhauser, Philipp Rentzsch, Stephen Krämer, Carolin Andresen, Jeongbin Park, Roland Eils, Matthias Schlesner, Carl Herrmann, ShinyButchR: interactive NMF-based decomposition workflow of genome-scale datasets, Biology Methods and Protocols, bpaa022.
ShinyButchR
We also provide an interactive R/Shiny app ShinyButchR to perform NMF and
explore the results interactively.
The live version can be used here:
https://hdsu-bioquant.shinyapps.io/shinyButchR/
Or a pre-build image can be pulled from Docker: https://hub.docker.com/r/hdsu/shinybutchr
docker run --rm -p 3838:3838 hdsu/shinybutchr
Introduction
NMF (nonnegative matrix factorization) is a matrix decomposition method. A description of the algorithm and it’s implementation can be found e.g. in (Lee and Seung 1999). In 2003, Brunet et al. applied NMF to gene expression data (Brunet et al. 2003). In 2010, NMF, an R package implementing several NMF solvers was published (Gaujoux and Seoighe 2010). NMF basically solves the problem as illustrated in the following figure (Image taken from https://en.wikipedia.org/wiki/Non-negative_matrix_factorization):
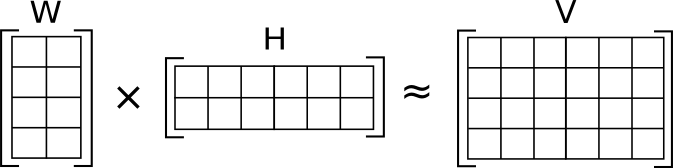
Here, V is an input matrix with dimensions n × m. It is decomposed into two matrices W of dimension n × l and H of dimension l × m, which when multiplied approximate the original matrix V. l is a free parameter in NMF, it is called the factorization rank. If we call the columns of W , then l corresponds to the number of signatures. The decomposition thus leads to a reduction in complexity if l < n, i.e. if the number of signatures is smaller than the number of features, as indicated in the above figure.
In 2015, Mejia-Roa et al. introduced an implementation of an NMF-solver in CUDA, which lead to significant reduction of computation times by making use of massive parallelisation on GPUs (Mejia-Roa et al. 2015). Other implementations of NMF-solvers on GPUs exist.
It is the pupose of the package ButchR described here to provide
wrapper functions in R to these NMF-solvers in TensorFlow. Massive
parallelisation not only leads to faster algorithms, but also makes the
benefits of NMF accessible to much bigger matrices. Furthermore,
functions for estimation of the optimal factorization rank and post-hoc
feature selection are provided.
The ButchR package
The matrix decomposition results are stored in an S4 object called
ButchR_NMF. ButchR provides functions to access the best
factorzation after n initailization W and H matrices for a given
factorzation rank.
NMF example - STAGE 1 preprocessing
For the tutorial we will use an RNA-seq dataset from labeled cell types of the human hematopoietic system, originally published on:
Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R, Chang HY. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203 (2016). https://doi.org/10.1038/ng.3646
In this session we will download and pre-process the data. The data will be downloaded as a counts matrix from GEO.
Preprocessing
Load required packages:
library(tidyverse)
library(viridis)
library(ComplexHeatmap)
library(DESeq2)
library(ButchR)
library(ggupset)
library(clusterProfiler)
library(msigdbr)
library(umap)
library(cowplot)
Download counts from GEO, select samples, and normalize data.
##----------------------------------------------------------------------------##
## Download counts ##
##----------------------------------------------------------------------------##
# set ftp url to RNA-seq data
ftp_url <- file.path("ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE74nnn/GSE74246",
"suppl/GSE74246_RNAseq_All_Counts.txt.gz")
read_delim_gz <- function(file_url) {
con <- gzcon(url(file_url))
txt <- readLines(con)
return(read.delim(textConnection(txt), row.names = 1))
}
# read in data matrix
corces_rna_counts <- read_delim_gz(ftp_url)
##----------------------------------------------------------------------------##
## Data loading and sample QC ##
##----------------------------------------------------------------------------##
corces_rna_counts[1:5,1:5]
Click for Answer
## X5852.HSC X6792.HSC X7256.HSC X7653.HSC X5852.MPP
## A1BG 14 9 1 5 13
## A1BG-AS1 3 0 1 0 27
## A1CF 0 0 0 0 0
## A2M 78 192 36 82 66
## A2M-AS1 71 76 52 86 49
dim(corces_rna_counts)
## [1] 25498 81
Remove leukemic and erythroblast samples:
corces_rna_counts <- corces_rna_counts[,-grep("Ery|rHSC|LSC|Blast", colnames(corces_rna_counts))]
dim(corces_rna_counts)
Click for Answer
## [1] 25498 46
Inspect correlation matrix:
cor_dm <- cor(corces_rna_counts)
Heatmap(cor_dm, col = magma(100), name = "Correlation")
Click for Answer
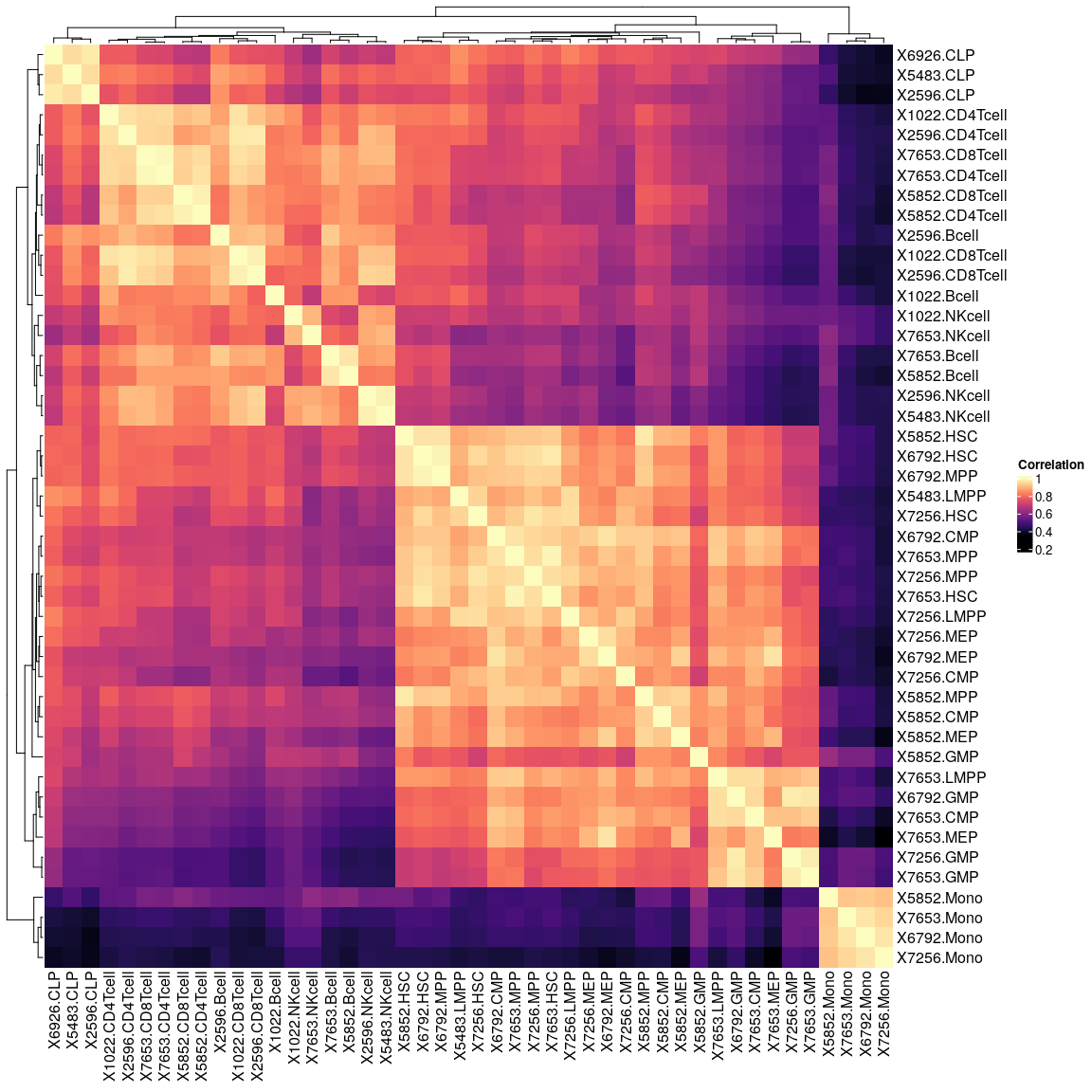
X5852.GMP is an outlier and will be removed, has much smaller library size as other GMPS:
rm(cor_dm)
corces_rna_counts <- corces_rna_counts[,-grep("X5852.GMP", colnames(corces_rna_counts))]
Remove rows with rowSum==0:
corces_rna_counts <- corces_rna_counts[!rowSums(corces_rna_counts) == 0,]
Normalize counts using DESeq2:
##----------------------------------------------------------------------------##
## Normalize counts ##
##----------------------------------------------------------------------------##
# do DESeq2 size factor normalization
sf <- estimateSizeFactorsForMatrix(corces_rna_counts)
corces_rna_counts <- t( t(corces_rna_counts) / sf )
# do +1 log2 transformation
corces_rna_norm <- apply(corces_rna_counts + 1, 2, log2)
rm(ftp_url, sf)
Format annotation data frame for future reference:
##----------------------------------------------------------------------------##
## Annotation ##
##----------------------------------------------------------------------------##
# extract celltypes from colnames
col.anno <- gsub(".*\\.", "", colnames(corces_rna_norm))
col.anno[grep("NK", col.anno)] <- "NK"
col.anno[grep("CD4", col.anno)] <- "CD4"
col.anno[grep("CD8", col.anno)] <- "CD8"
# Define color vector
type.color <- setNames(c("#771155", "#AA4488", "#CC99BB", "#114477", "#4477AA", "#77AADD",
"#117777", "#44AAAA", "#77CCCC", "#777711", "#AAAA44", "#DDDD77"),
c("HSC", "MPP", "LMPP", "CMP", "GMP", "MEP",
"CLP", "CD4", "CD8", "NK", "Bcell", "Mono"))
# Annotation data frame
corces_rna_annot <- data.frame(sampleID = colnames(corces_rna_norm),
Celltype = as.factor(col.anno),
color = type.color[match(col.anno, names(type.color))],
row.names = colnames(corces_rna_norm),
stringsAsFactors = FALSE)
Dimension of transcriptome dataset (RNA-seq):
dim(corces_rna_norm)
Click for Answer
## [1] 21811 45
Preprocessing steps for the Corces 2016 ATAC-seq and to run iNMF can be found here