[BC]2 Tutorial
Defining genomic signatures with Non-Negative Matrix Factorization
Carl Herrmann & Andres Quintero
13 September 2021Non-Negative Matrix factorization example - STAGE 1 preprocessing
For the tutorial we will use RNA-seq and ATAC-seq datasets from labeled cell types of the human hematopoietic system, originally published on:
Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R, Chang HY. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203 (2016). https://doi.org/10.1038/ng.3646
In this session we will download and pre-process the data. The data will be downloaded as counts matrices from GEO.
Process RNAseq data
Download and read RNAseq data.
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Set paths ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
rna.data.path <- "data/rnaseq/"
GSE74246.path <- file.path(rna.data.path, "GSE74246_RNAseq_All_Counts.txt.gz")
dir.create(rna.data.path, recursive = TRUE, showWarnings = FALSE)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Download counts ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# set ftp url to RNA-seq data
ftp.url <- file.path("ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE74nnn/GSE74246",
"/suppl/GSE74246_RNAseq_All_Counts.txt.gz")
# download data
if(!file.exists(GSE74246.path)){
download.file(url = ftp.url, GSE74246.path)
}
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Data loading and sample QC ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# read in data matrix
rna.counts <- read.delim(gzfile(GSE74246.path), row.names = 1)
rna.counts[1:5,1:5]
Click for Answer
## X5852.HSC X6792.HSC X7256.HSC X7653.HSC X5852.MPP
## A1BG 14 9 1 5 13
## A1BG-AS1 3 0 1 0 27
## A1CF 0 0 0 0 0
## A2M 78 192 36 82 66
## A2M-AS1 71 76 52 86 49
dim(rna.counts)
## [1] 25498 81
Remove leukemic and erythroblast samples.
rna.counts <- rna.counts[,-grep("Ery|rHSC|LSC|Blast", colnames(rna.counts))]
dim(rna.counts)
Click for Answer
## [1] 25498 46
Inspect correlation matrix.
cor.dm <- cor(rna.counts)
Heatmap(cor.dm, col = magma(100), name = "Correlation")
Click for Answer
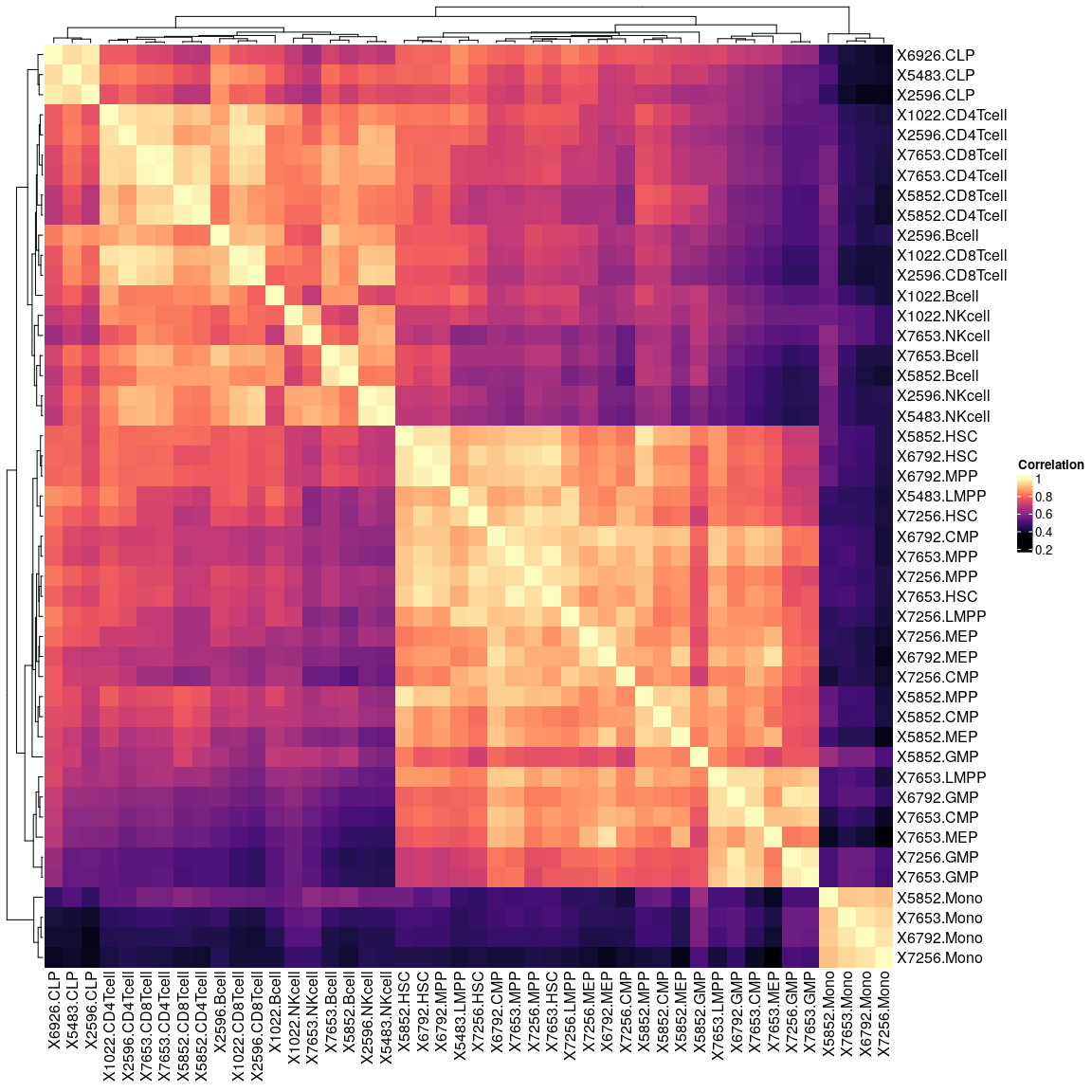
X5852.GMP is an outlier and will be removed, has much smaller library size as other GMPS.
Also remove genes with 0 counts.
rna.counts <- rna.counts[,-grep("X5852.GMP", colnames(rna.counts))]
# remove rows with rowSum==0
rna.counts <- rna.counts[!rowSums(rna.counts) == 0,]
Normalize counts and format annotation data frame.
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Normalize counts ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# do DESeq2 size factor normalization
sf <- estimateSizeFactorsForMatrix(rna.counts)
rna.counts <- t( t(rna.counts) / sf )
# do +1 log2 transformation
rna.norm.mat <- apply(rna.counts + 1, 2, log2)
rm(ftp.url, GSE74246.path, sf)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Annotation ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# extract celltypes from colnames
col.anno <- gsub(".*\\.", "", colnames(rna.norm.mat))
col.anno[grep("NK", col.anno)] <- "NK"
col.anno[grep("CD4", col.anno)] <- "CD4"
col.anno[grep("CD8", col.anno)] <- "CD8"
# Define color vector
type.color <- setNames(c("#771155", "#AA4488", "#CC99BB", "#114477", "#4477AA", "#77AADD",
"#117777", "#44AAAA", "#77CCCC", "#777711", "#AAAA44", "#DDDD77"),
c("HSC", "MPP", "LMPP", "CMP", "GMP", "MEP",
"CLP", "CD4", "CD8", "NK", "Bcell", "Mono"))
# Annotation data frame
rna.annot <- data.frame(sampleID = colnames(rna.norm.mat),
Celltype = as.factor(col.anno),
color = type.color[match(col.anno, names(type.color))],
row.names = colnames(rna.norm.mat),
stringsAsFactors = FALSE)
Save data
saveRDS(rna.norm.mat, "data/rnaseq/rnaseq_normalized_counts.RDS")
saveRDS(rna.annot, "data/rnaseq/rnaseq_annotation.RDS")
Process ATACseq data
Download and read RNAseq data.
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Set paths ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
atac.data.path <- file.path("data/atacseq/")
GSE74912.path <- file.path(atac.data.path, "GSE74912_ATACseq_All_Counts.txt.gz")
dir.create(atac.data.path, recursive = TRUE, showWarnings = FALSE)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Download counts ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# download data
if(!file.exists(GSE74912.path)){
download.file(url = "ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE74nnn/GSE74912/suppl/GSE74912_ATACseq_All_Counts.txt.gz", GSE74912.path)
}
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Data loading and sample QC ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# read in data matrix
atac.counts <- read.delim(gzfile(GSE74912.path), stringsAsFactors = FALSE)
atac.counts[1:5,1:5]
Click for Answer
## Chr Start End X4983.1A X4983.2A
## 1 chr1 10025 10525 1 0
## 2 chr1 13252 13752 0 5
## 3 chr1 16019 16519 4 2
## 4 chr1 96376 96876 0 0
## 5 chr1 115440 115940 0 3
Separate annotation column from data.
atac.row.anno <- atac.counts[,1:3]
atac.counts <- atac.counts[,-c(1:3)]
dim(atac.counts)
Click for Answer
## [1] 590650 132
Remove rows with rowSums < 2000 and leukemic and erythroblast samples.
rownames(atac.counts) <- do.call(paste, c(as.list(atac.row.anno), sep = "_"))
# remove rows with rowSums < 2000
atac.row.anno <- atac.row.anno[rowSums(atac.counts) > 2000,]
atac.counts <- atac.counts[rowSums(atac.counts) > 2000,]
# remove leukemic and erythroblast samples
atac.counts <- atac.counts[,-grep("Ery|LSC|pHSC|Leuk|CD34", colnames(atac.counts))]
dim(atac.counts)
Click for Answer
``` ## [1] 123102 69 ```sum(rowSums(atac.counts) == 0)
## [1] 0
Compute correlation matrix.
Heatmap(cor(atac.counts), col = magma(100), name = "Correlation")
Click for Answer
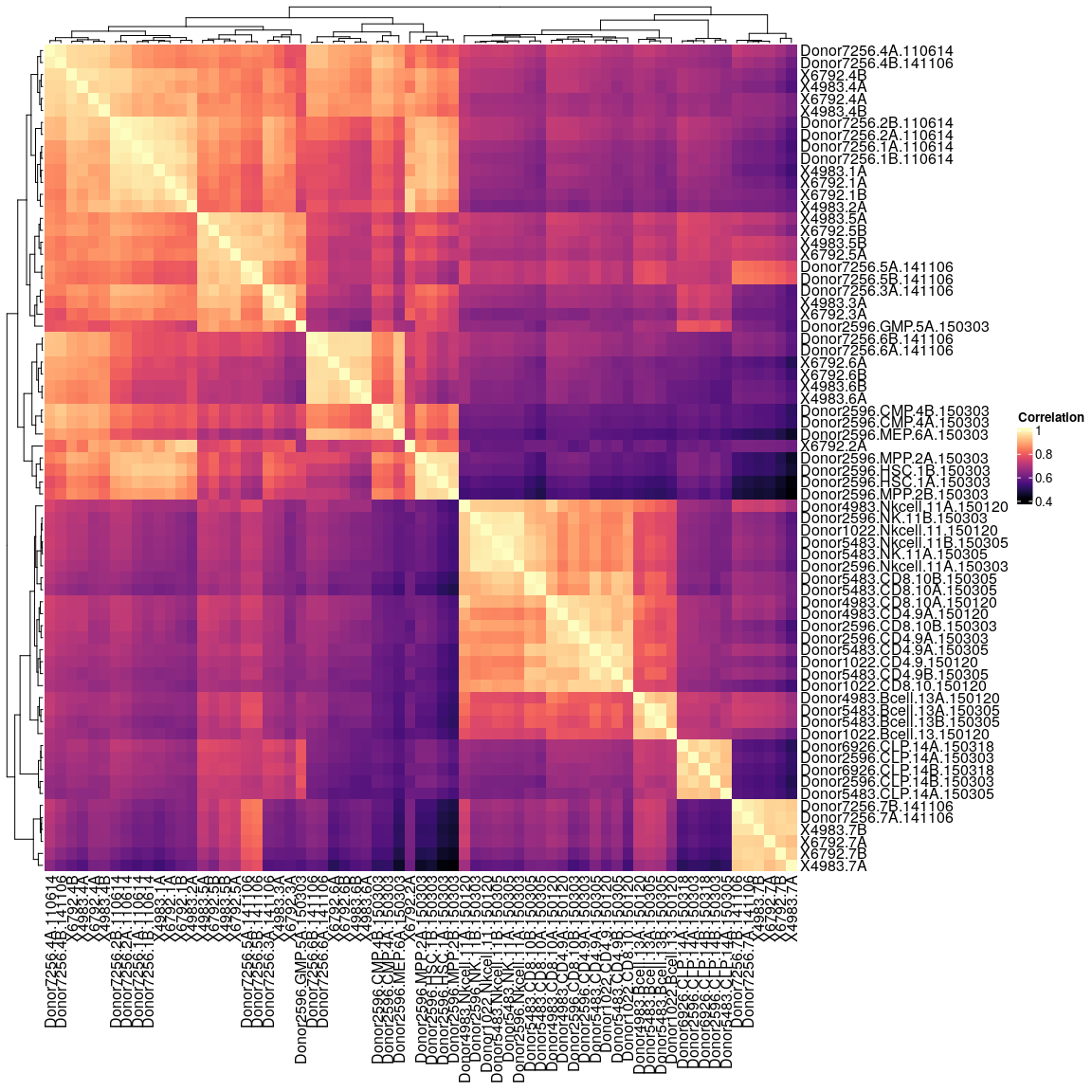
Remove X6792.7A, due to low coverage.
atac.counts <- atac.counts[,-grep("X6792.7A", colnames(atac.counts))]
Normalize counts and format annotation data frame.
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Normalize counts ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# do DESeq2 norm
sf <- estimateSizeFactorsForMatrix(atac.counts)
atac.counts <- t(t(atac.counts)/sf)
# transform to log2
atac.norm.mat <- apply(atac.counts + 1, 2, log2)
rm(sf, GSE74912.path)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Annotation ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# extract celltypes from colnames
col.anno <- colnames(atac.norm.mat)
col.anno[grep("CD4", col.anno)] <- "CD4"
col.anno[grep("CD8", col.anno)] <- "CD8"
col.anno[grep("NK", col.anno)] <- "NK"
col.anno[grep("Nkcell", col.anno)] <- "NK"
col.anno[grep("Bcell", col.anno)] <- "Bcell"
col.anno[grep("CLP", col.anno)] <- "CLP"
col.anno[grep("1(A|B)", col.anno)] <- "HSC"
col.anno[grep("2(A|B)", col.anno)] <- "MPP"
col.anno[grep("3(A|B)", col.anno)] <- "LMPP"
col.anno[grep("4(A|B)", col.anno)] <- "CMP"
col.anno[grep("5(A|B)", col.anno)] <- "GMP"
col.anno[grep("6(A|B)", col.anno)] <- "MEP"
col.anno[grep("7(A|B)", col.anno)] <- "Mono"
col.anno
Click for Answer
## [1] "HSC" "MPP" "LMPP" "CMP" "CMP" "GMP" "GMP" "MEP" "MEP"
## [10] "Mono" "Mono" "HSC" "HSC" "MPP" "LMPP" "CMP" "CMP" "GMP"
## [19] "GMP" "MEP" "MEP" "Mono" "Bcell" "CD4" "CD8" "NK" "CD4"
## [28] "CD8" "CLP" "CLP" "CMP" "CMP" "GMP" "HSC" "HSC" "MEP"
## [37] "MPP" "MPP" "NK" "NK" "Bcell" "CD4" "CD8" "NK" "Bcell"
## [46] "CD4" "CD4" "CD8" "CLP" "NK" "NK" "CLP" "HSC" "HSC"
## [55] "MPP" "MPP" "LMPP" "CMP" "CMP" "GMP" "GMP" "MEP" "MEP"
## [64] "Mono" "Mono" "Bcell" "CLP" "CD8"
# Annotation data frame
atac.annot <- data.frame(sampleID = colnames(atac.norm.mat),
Celltype = as.factor(col.anno),
color = type.color[match(col.anno, names(type.color))],
row.names = colnames(atac.norm.mat),
stringsAsFactors = FALSE)
Save data.
saveRDS(atac.norm.mat, "data/atacseq/atacseq_normalized_counts.RDS")
saveRDS(atac.annot, "data/atacseq/atacseq_annotation.RDS")
Match samples
Find samples with RNAseq and ATACseq data available.
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Match RNAseq and ATACseq ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## ATACseq annotations ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# replace numbered celltypes by character names
col.anno <- colnames(atac.counts)
col.anno[grep("CD4", col.anno)] <- "CD4"
col.anno[grep("CD8", col.anno)] <- "CD8"
col.anno[grep("NK", col.anno)] <- "NK"
col.anno[grep("Nkcell", col.anno)] <- "NK"
col.anno[grep("Bcell", col.anno)] <- "Bcell"
col.anno[grep("CLP", col.anno)] <- "CLP"
col.anno[grep("1(A|B)", col.anno)] <- "HSC"
col.anno[grep("2(A|B)", col.anno)] <- "MPP"
col.anno[grep("3(A|B)", col.anno)] <- "LMPP"
col.anno[grep("4(A|B)", col.anno)] <- "CMP"
col.anno[grep("5(A|B)", col.anno)] <- "GMP"
col.anno[grep("6(A|B)", col.anno)] <- "MEP"
col.anno[grep("7(A|B)", col.anno)] <- "Mono"
atac.anno.cellID <- col.anno
rm(col.anno)
# Paste donor ID and cell type
atac.anno <- paste0(sapply(strsplit(colnames(atac.counts), "\\."), "[[", 1), ".", atac.anno.cellID)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## RNAseq annotations ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# Keep only Donor ID
rnaseqIDs <- setNames(sapply(strsplit(colnames(rna.counts), "\\."), "[[", 1), colnames(rna.counts))
rnaseqIDs <- sub("^X", "", rnaseqIDs)
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Match RNAseq and ATACseq ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
rna.atac.matched.samples <- lapply(setNames(1:length(rnaseqIDs), names(rnaseqIDs)), function(i) {
# Find same donor in RNAseq and ATACseq
atac.matched <- grep(rnaseqIDs[i], atac.anno, value = TRUE)
if (length(atac.matched) > 0) {
# if same donor in both omics search if same cell type
# extract cell tupe fron RNAseq colname
anno <- sapply(strsplit(names(rnaseqIDs)[i], "\\."), "[[", 2)
anno <- sub("Tcell", "", anno)
anno <- sub("cell", "", anno)
# find in ATACseq
anno.matched <- grep(anno, atac.matched, value = TRUE)
if (anno == "MPP") {
anno.matched <- grep("LMPP", anno.matched, value = TRUE, invert = TRUE)
}
if (length(anno.matched) > 0) {
data.frame(rnaID = names(rnaseqIDs)[i],
atacID = colnames(atac.counts)[atac.anno %in% anno.matched],
cellID = atac.anno.cellID[atac.anno %in% anno.matched],
atac.anno = anno.matched,
row.names = colnames(atac.counts)[atac.anno %in% anno.matched])
}
}
} )
# Keep only matched sampels
sum(!sapply(rna.atac.matched.samples, is.null))
Click for Answer
## [1] 24
rna.atac.matched.samples <- rna.atac.matched.samples[!sapply(rna.atac.matched.samples, is.null)]
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Match RNAseq and ATACseq ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# Keep only one ATACseq replicate
rna.atac.annot <- do.call(rbind, rna.atac.matched.samples)
rna.atac.annot <- rna.atac.annot[!duplicated(rna.atac.annot$rnaID),]
# Subset intersect on multi views
multiview.norm.mat <- list(rna = rna.norm.mat[, match(rna.atac.annot$rnaID, colnames(rna.norm.mat))],
atac = atac.norm.mat[, match(rna.atac.annot$atacID, colnames(atac.norm.mat))])
lapply(multiview.norm.mat, dim)
Click for Answer
## $rna
## [1] 21811 24
##
## $atac
## [1] 123102 24
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Format multi view to easy access ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
# Format Colnames
lapply(multiview.norm.mat, colnames)
Click for Answer
## $rna
## [1] "X6792.HSC" "X7256.HSC" "X6792.MPP" "X7256.MPP"
## [5] "X7256.LMPP" "X6792.CMP" "X7256.CMP" "X6792.GMP"
## [9] "X7256.GMP" "X6792.MEP" "X7256.MEP" "X6792.Mono"
## [13] "X7256.Mono" "X1022.CD4Tcell" "X2596.CD4Tcell" "X1022.CD8Tcell"
## [17] "X2596.CD8Tcell" "X1022.NKcell" "X2596.NKcell" "X5483.NKcell"
## [21] "X1022.Bcell" "X2596.CLP" "X5483.CLP" "X6926.CLP"
##
## $atac
## [1] "X6792.1A" "Donor7256.1A.110614"
## [3] "X6792.2A" "Donor7256.2A.110614"
## [5] "Donor7256.3A.141106" "X6792.4A"
## [7] "Donor7256.4A.110614" "X6792.5A"
## [9] "Donor7256.5A.141106" "X6792.6A"
## [11] "Donor7256.6A.141106" "X6792.7B"
## [13] "Donor7256.7A.141106" "Donor1022.CD4.9.150120"
## [15] "Donor2596.CD4.9A.150303" "Donor1022.CD8.10.150120"
## [17] "Donor2596.CD8.10B.150303" "Donor1022.Nkcell.11.150120"
## [19] "Donor2596.NK.11B.150303" "Donor5483.NK.11A.150305"
## [21] "Donor1022.Bcell.13.150120" "Donor2596.CLP.14A.150303"
## [23] "Donor5483.CLP.14A.150305" "Donor6926.CLP.14A.150318"
colnames(multiview.norm.mat$atac) <- colnames(multiview.norm.mat$rna)
lapply(multiview.norm.mat, colnames)
Click for Answer
## $rna
## [1] "X6792.HSC" "X7256.HSC" "X6792.MPP" "X7256.MPP"
## [5] "X7256.LMPP" "X6792.CMP" "X7256.CMP" "X6792.GMP"
## [9] "X7256.GMP" "X6792.MEP" "X7256.MEP" "X6792.Mono"
## [13] "X7256.Mono" "X1022.CD4Tcell" "X2596.CD4Tcell" "X1022.CD8Tcell"
## [17] "X2596.CD8Tcell" "X1022.NKcell" "X2596.NKcell" "X5483.NKcell"
## [21] "X1022.Bcell" "X2596.CLP" "X5483.CLP" "X6926.CLP"
##
## $atac
## [1] "X6792.HSC" "X7256.HSC" "X6792.MPP" "X7256.MPP"
## [5] "X7256.LMPP" "X6792.CMP" "X7256.CMP" "X6792.GMP"
## [9] "X7256.GMP" "X6792.MEP" "X7256.MEP" "X6792.Mono"
## [13] "X7256.Mono" "X1022.CD4Tcell" "X2596.CD4Tcell" "X1022.CD8Tcell"
## [17] "X2596.CD8Tcell" "X1022.NKcell" "X2596.NKcell" "X5483.NKcell"
## [21] "X1022.Bcell" "X2596.CLP" "X5483.CLP" "X6926.CLP"
# Format annotation
rna.atac.annot$original.atacID <- rna.atac.annot$atacID
rna.atac.annot$atacID <- rna.atac.annot$rnaID
rownames(rna.atac.annot) <- rna.atac.annot$rnaID
rna.atac.annot <- rna.atac.annot[,c(2,3,1,4,5)]
colnames(rna.atac.annot) <- c("sampleID", "Celltype", "rna.sampleID", "atac.sampleID", "original.atacID")
rna.atac.annot$color <- type.color[match(rna.atac.annot$Celltype, names(type.color))]
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
## Save data ##
##––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––##
dir.create("data/multiview/", recursive = TRUE)
# Save annotation
saveRDS(rna.atac.annot, file = "data/multiview/multiview_annotation.RDS")
write.table(rna.atac.annot, "data/multiview/multiview_annotation.csv", quote = FALSE, sep = "\t", row.names = FALSE)
# Save normalized matrices
saveRDS(multiview.norm.mat, file = "data/multiview/multiview_norm_mat_list.RDS")