[BC]2 Tutorial
Defining genomic signatures with Non-Negative Matrix Factorization
Carl Herrmann & Andres Quintero
13 September 2021Test ButcherR-[BC]2 Docker image
After running the ButcherR-[BC]2 Docker image (see Run Docker image hdsu/butcher-bc2), you can run the following short example with a small leukemia dataset to test it.
Step 1 - Sign in to Rstudio
With the app open, sign in to RStudio using the following credentials:
- Username: hdsu
- Password: pass
Click for Answer
 |
|---|
| To start use "hdsu" as username and "pass" as password |
 |
|---|
| Screen you should see after logging in to RStudio |
Step 2 - Load packages
Run the following lines to load the required packages:
library(ButchR)
library(ComplexHeatmap)
library(viridis)
library(tidyverse)
Step 3 - Load leukemia data
Load the example data
data(leukemia)
Now we are ready to start an NMF analysis.
Step 4 - run NMF
The wrapper function for the NMF solvers in the ButchR package is
run_NMF_tensor. It is called as follows:
k_min <- 2
k_max <- 4
leukemia_nmf_exp <- run_NMF_tensor(X = leukemia$matrix,
ranks = k_min:k_max,
method = "NMF",
n_initializations = 10,
extract_features = TRUE)
Click for Answer
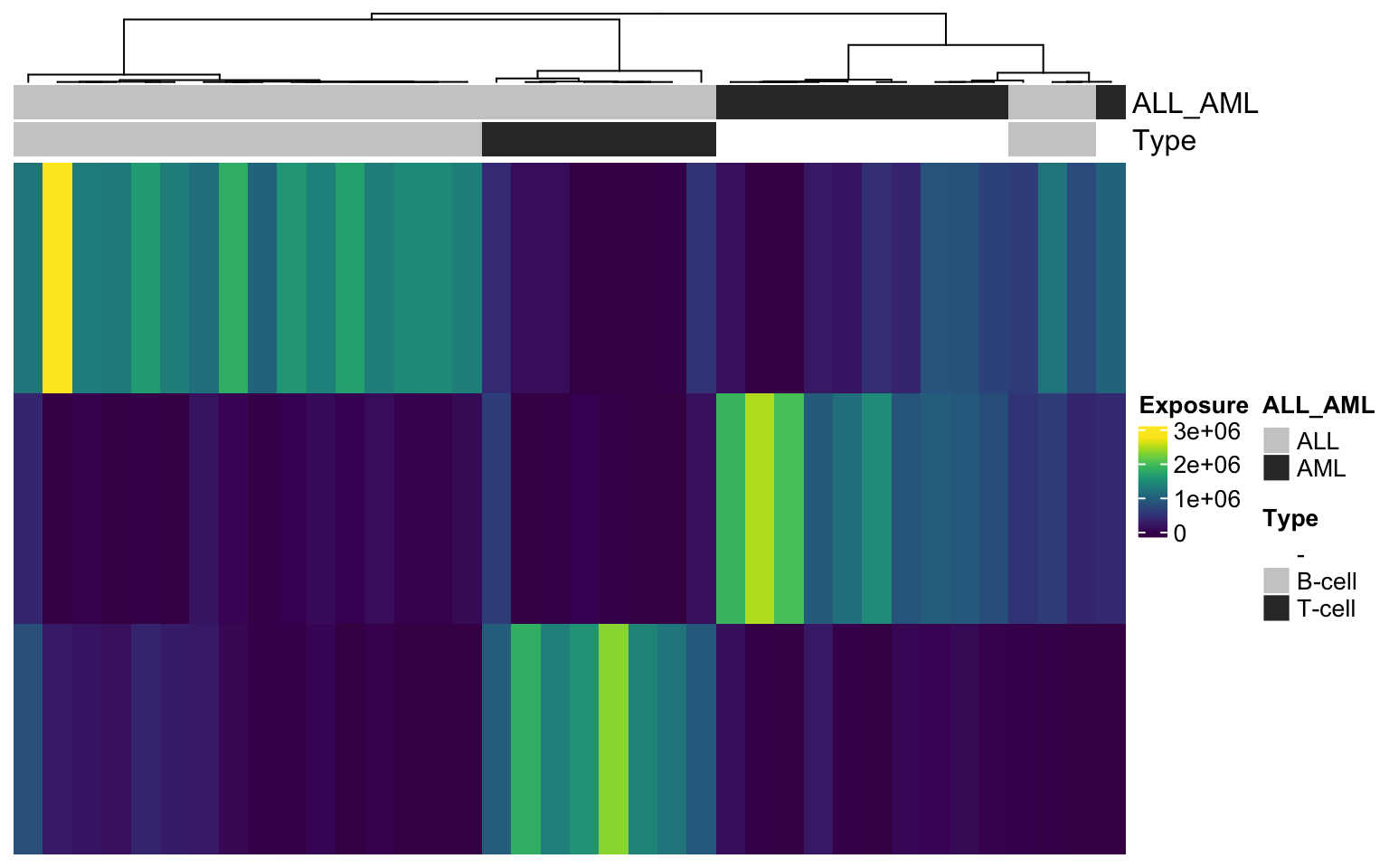
H matrix for k= 3
## [1] "2020-07-16 17:50:42 CEST"
## Factorization rank: 2
## [1] "NMF converged after 75,123,64,69,58,126,141,83,54,87 iterations"
## [1] "2020-07-16 17:50:42 CEST"
## Factorization rank: 3
## [1] "NMF converged after 154,79,90,87,66,84,76,151,115,102 iterations"
## [1] "2020-07-16 17:50:44 CEST"
## Factorization rank: 4
## [1] "NMF converged after 108,189,202,108,121,76,104,150,110,132 iterations"
## No optimal K could be determined from the Optimal K stat
Depending on the choice of parameters (dimensions of the input matrix, number of iterations), this step may take some time. Note that the algorithm updates the user about the progress in the iterations.
Step 5 - Normalize W matrix
To make the features in the W matrix comparable, the factorization is normalized to make all columns of W sum 1.
leukemia_nmf_exp <- normalizeW(leukemia_nmf_exp)
Step 6 - Visualize the matrix H (exposures)
The matrices H may be visualized as heatmaps. We can define a meta
information object and annotate meta data:
heat_anno <- HeatmapAnnotation(df = leukemia$annotation[, c("ALL_AML", "Type")],
col = list(ALL_AML = c("ALL" = "grey80",
"AML" = "grey20"),
Type = c("-" = "white",
"B-cell" = "grey80",
"T-cell" = "grey20")))
And now display the matrix H with meta data annotation:
#plot H matrix
tmp_hmatrix <- HMatrix(leukemia_nmf_exp, k = 3)
Heatmap(tmp_hmatrix,
col = viridis(100),
name = "Exposure",
clustering_distance_columns = 'pearson',
show_column_dend = TRUE,
top_annotation = heat_anno,
show_column_names = FALSE,
show_row_names = FALSE,
cluster_rows = FALSE)
Click for Answer
H matrix for k= 3