IRTG Course
Introduction to R for genomics
Carl Herrmann & Carlos Ramirez
8-9 December 2021Quality control
Filtering cells with low sequencing quality is a very important step since it can greatly impact in further analysis. Quality check and control often requires to visualize and inspect samples in order to determine appropiate thresholds. Threshold values might vary from one dataset to another, so no hard threshold rule can be applied equally to every case.
We will examine the number of UMI counts, the number of RNA features and the percentage of reads of mitochondrial genes.
We will first calculate the percentage of UMI counts of reads mapped to mitochondrial genes. This
step most be manually done since is based on a priori knowledge of which genes corresponds to
mitochondrial genes.
In this case, genes are annotated using human ensembl gene symbol annotations mitochondrial
genes are annotated starting with a MT- string.
pbmc.seurat[["percent.mt"]] <- PercentageFeatureSet(pbmc.seurat, pattern = "^MT-")
Then, we can visualize the following metrics.
-
Number of features - Correspond to the number of different mapped genomic features. For example, in the case of scRNA-Seq features corresponds to genes, in ATAC-Seq to genomic ranges, etc. High number of features can indicate doublets and empty cells. Usually between 1 and 30000.
-
Number of counts - Number of mapped reads. It can also indicate the presence of doublets and empty droplet. It's generally correlated to the number of features. Usually between 1 and 20000.
-
Percentage of mitochondrial genes - Percentage of mapped reads that are annotated to mitochondrial genes. The presence of high levels of % of mitochondrial genes can suggest that a cell have lost its membrane integrity or that the cytoplasm has been leaked off and only the mitochondria was retained. Usually a range from 0 to 10 is aceptable.
We can plot these metrics using the function VlnPlot() as follows:
VlnPlot(pbmc.seurat, features = c('nFeature_RNA', 'nCount_RNA', 'percent.mt'))
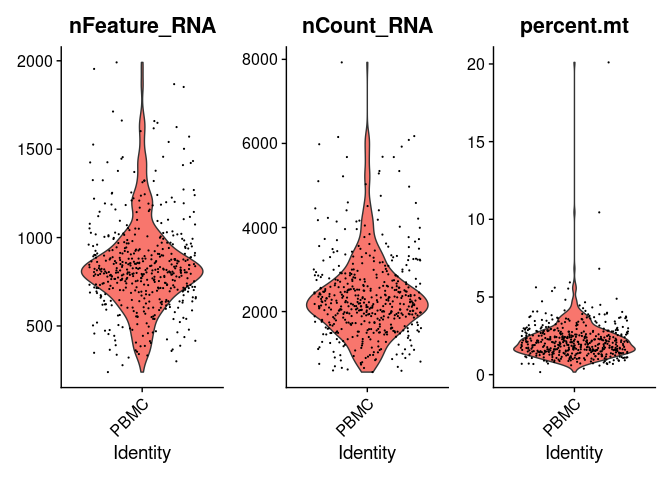
The violin plots show the values of the metrics for each cell along with an adjusted violin distribution.
Filtering out cells
Based on the previous violin plots we can define some thresholds and filter out cells using
the subset function. In the following code we select cells having nFeatures < 1250,
nCount < 4000 and percentage of reads mapped to mitochondrial genes < 5 percent.
pbmc.filtered <- subset(pbmc.seurat,
nFeature_RNA < 1250 &
nCount_RNA < 4000 &
percent.mt < 5)
We can check the number of cell that passed the QC.
ncol(pbmc.filtered)
## [1] 452
Normalization
There are several methods for normalization of scRNA-Seq data. A commonly used strategy is the log normalization which basically corrects sequencing deep in cells by dividing each feature by the total number of counts and then multiplied the result by a factor, usually 10000, and finally the values are log transformed.
Log normalization can be implemented by using the NormalizeData() function.
pbmc.filtered <- NormalizeData(pbmc.filtered)
Then, in order to make genes measurements more comparable log transformed values are scaled in a way that the media is equal to zero and the variance is equal to 1 as follows:
pbmc.filtered <- ScaleData(pbmc.filtered)
Quizzes
Performing your own QC!
QUIZ 1
How are the number of features and UMI counts related?
a) They are not related and randomly distributed in a scatter plot
b) They are related in a non-linear way
c) They are linearly related
TIP: Use the function FeatureScatter, inspect the manual using ?function.
Answer:
FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
We observe as expected a linear relation between the number of UMI counts and the features recorded.
QUIZ 2
Load a seurat object using the following command:
Warning!! Check your Seurat version, and use one of the two commands:
- if you have Seurat version 4.xx:
pbmc.seurat <- readRDS(url('https://raw.githubusercontent.com/caramirezal/caramirezal.github.io/master/bookdown-minimal/data/pbmc_10X_250_cells.seu.rds'))
- if you have Seurat version 3.xx:
pbmc.seurat <- readRDS(url('https://www.dropbox.com/s/pdeaq8rgzp86lyv/pbmc_10X_200_cells.seu.rds?dl=1'))
*Calculate the mean and median values of the percentage of mitochondrial reads*
- mean=2.2133 and median=2.0532
- mean=2.2246 and median=2.0639
- mean=0.0102. and median=1.1743
Filter out cells based on QC values
NOTE: Do not skip any step in the pipeline.