Workshop ChIPATAC 2020
Computational analysis of ChIP-seq and ATAC-seq data
14-15 December 20205. ChIP-seq : Peak Calling
Once the IP and control libraries have been aligned, and the aligned bam files filtered for low-quality reads, we want to determine the regions in which the IP signal is significantly higher than the control (=background) signal. This is called peak calling. There are many peak calling tools available, which are based on different statistical assumptions. We will use a widely used too called MACS2.
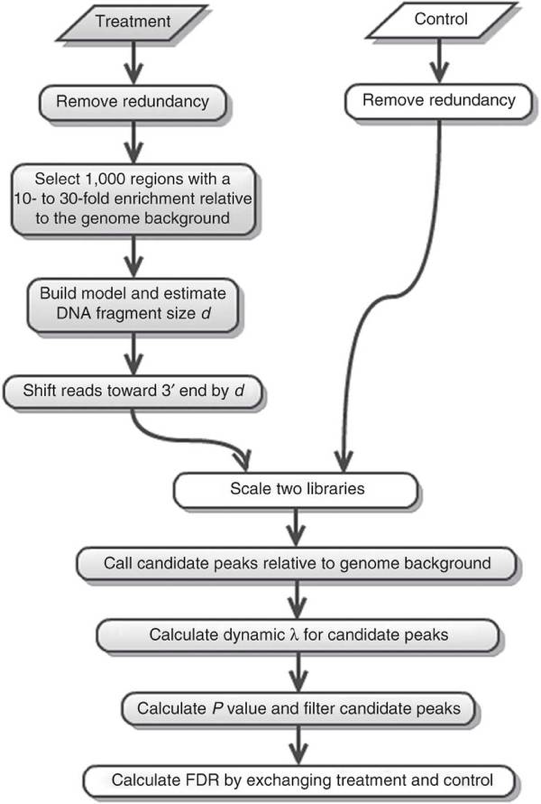
The workflow is the following:
Narrow CTCF peak
# Go to your home directory
cd
# Create a folder for your analysis
mkdir -p analysis/MACS2/CTCF
# Check out all the available parameters in MACS2
# Do note, when in doubt, its often good practice to use default settings
# Most options are optional and set to default, focus on the essential parameters that has to be changed
macs2 --help
# Launch the MACS2 peak calling
macs2 callpeak \
--treatment data/processed/CTCF/Bowtie2/CTCF_Rep1_ENCFF001HLV_trimmed_aligned_filt_sort_nodup.bam \
--control data/processed/CTCF/Bowtie2/CTCF_Control_ENCFF001HME_trimmed_aligned_filt_sort_nodup.bam \
--name CTCF \
--format BAM \
--keep-dup all \
--gsize 2.7e9 \
--qvalue 0.01 \
--outdir analysis/MACS2/CTCF
You can find all generated files in the analysis/MACS2/CTCF folder.
Broad H3K4me3 peak
# Go to your home directory
cd
# Create a folder for your analysis
mkdir -p analysis/MACS2/H3K4me3
# Check out all the available parameters in MACS2
# Do note, when in doubt, its often good practice to use default settings
# Most options are optional and set to default, focus on the essential parameters that has to be changed
macs2 --help
macs2 callpeak \
--treatment data/processed/H3K4me3/Bowtie2/H3K4me3_Rep1_ENCFF001FIS_aligned_filt_sort_nodup.bam \
--control data/processed/H3K4me3/Bowtie2/H3K4me3_Control_ENCFF001HME_aligned_filt_sort_nodup.bam \
--name H3K4me3 \
--format BAM \
--keep-dup all \
--gsize 2.7e9 \
--broad \
--qvalue 0.05 \
--broad-cutoff 0.05 \
--outdir analysis/MACS2/H3K4me3
Analyze the outputs from MACS2, download the peaks
.xlsfiles using Cyberduck and check its contents; how many peaks do you get for each ChIP dataset?
From the output of MACS2 on the screen, what is the infered fragment length?
Regarding this last point, MACS2 also generates an R script, which we can execute, in order to better understand the model built by MACS2:
cd
/usr/bin/Rscript analysis/MACS2/CTCF/CTCF_model.r
/usr/bin/Rscript analysis/MACS2/H3K4me3/H3K4me3_model.r
Using Cyberduck, look at the pdf files generated in your home folder.
Do you understand these plots?
Run the H3K4me3 peak calling without the
--broadand--broad-cutoffoption; i.e. in narrow peak finding mode. Do you get more/less peaks?
Can you think of a reason why we keep
--keep-dup alloption ? Hint: think about the filtering steps used during alignment.
[Click to show answer!]
We have already removed all duplicate reads using the samtools markdup command, hence there are no duplicates left in the file! Without this preliminary filtering, we could have used the auto option to let MACS2 decide on a maximal number of duopliactes to keep!