[BC]2 Tutorial
Defining genomic signatures with Non-Negative Matrix Factorization
Carl Herrmann & Andres Quintero
13 September 2021Feature extraction and enrichment analysis
Identification of signature specific features
ButchR has a complete suite of functions to identify the differential contribution of a feature to every signature, classifying them into signature specific features and multi-signature features.
The function SignatureSpecificFeatures classifies one feature as non-contributing or contributing towards a signature definition, returning a binary matrix of features, where 0=non-contributing and 1=contributing.
ss_features <- SignatureSpecificFeatures(rna_norm_nmf_exp, k = 8,return_all_features = TRUE)
colnames(ss_features) <- paste0("Sig", 1:8)
ssf_gg <- ss_features %>%
as_tibble(rownames = "geneID") %>%
pivot_longer(cols = -geneID, names_to = "SigID", values_to = "IsSig") %>%
filter(IsSig == 1 ) %>%
dplyr::select(-IsSig ) %>%
group_by(geneID) %>%
summarize(SigID = list(SigID)) %>%
ggplot(aes(x = SigID)) +
geom_bar(fill=c(rep("red",8),rep("black",12))) +
scale_x_upset(order_by = "degree", n_intersections = 20) +
cowplot::theme_cowplot()
ssf_gg
Click for Answer
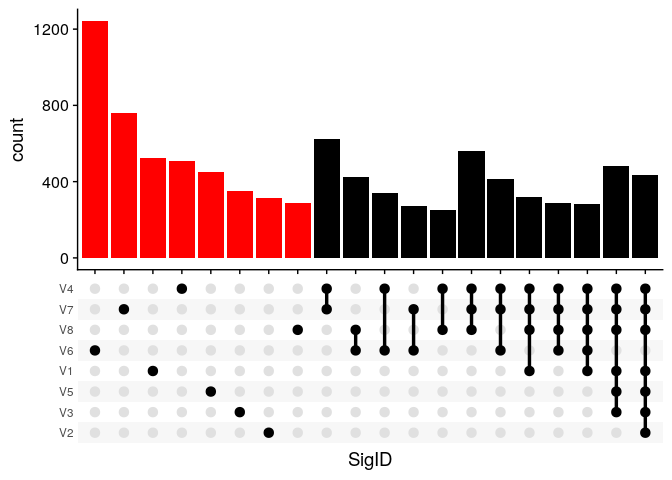
Ectract top signature specfic features
Notice that when the return_all_features flag is not used, the function SignatureSpecificFeatures returns a list of associated features with every signature.
Visual inspection of the top 10% of the signature specific features (i.e., signature specific genes):
ss_features <- SignatureSpecificFeatures(rna_norm_nmf_exp, k = 8)
ss_features <- do.call(c, ss_features)
wmatrix_norm <- WMatrix(rna_norm_nmf_exp, k = 8)
colnames(wmatrix_norm) <- paste0("Sig", 1:8)
##----------------------------------------------------------------------------##
## top 10% features Heatmap ##
##----------------------------------------------------------------------------##
top_10perc_assing <- function(wmatrix){
sig_assign <- lapply(setNames(colnames(wmatrix), colnames(wmatrix)), function(sigID){
selec_wmatrix <- do.call(cbind, lapply(as.data.frame(wmatrix), function(sign_expo){
sign_expo[sign_expo < quantile(sign_expo, 0.9)] <- NA
sign_expo
}))
rownames(selec_wmatrix) <- rownames(wmatrix)
selec_wmatrix <- selec_wmatrix[!is.na(selec_wmatrix[,sigID]),,drop=FALSE]
# Keep only the top feature if there's an overlap
sig_SE_IDs <- rownames(selec_wmatrix[rowMaxs(selec_wmatrix, na.rm = TRUE) == selec_wmatrix[,sigID],])
sig_SE_IDs
})
sig_assign
}
sign_features <- top_10perc_assing(wmatrix_norm)
wmatrix_norm_sel <- wmatrix_norm[do.call(c, sign_features), ]
dim(wmatrix_norm_sel)
Click for Answer
## [1] 7859 8
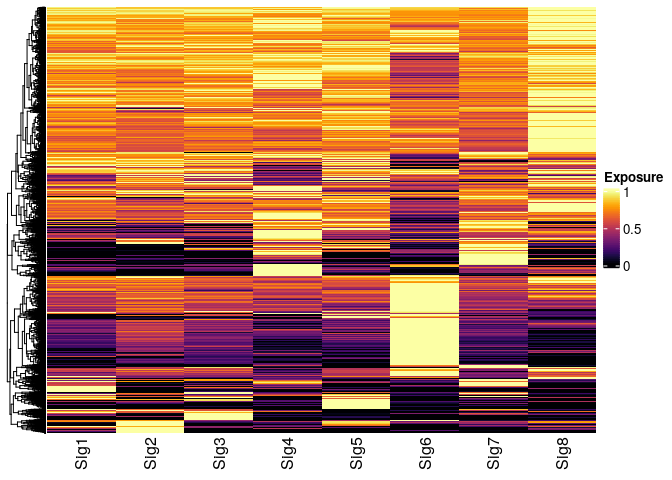
Heatmap(wmatrix_norm_sel/rowMaxs(wmatrix_norm_sel),
col = inferno(100),
name = "Exposure",
show_row_names = FALSE,
cluster_columns = FALSE )
Click for Answer
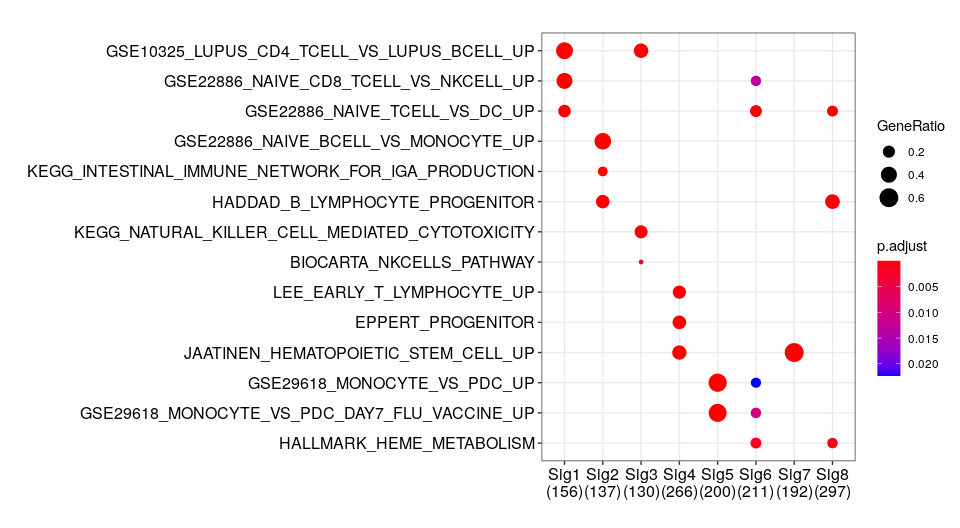
Gene set enrichment analysis of signature specific features
Gene set enrichment analysis using the same set of genes displayed in the previous heatmap. -log10 of the corrected p-values are shown for representative gene set collections.
##----------------------------------------------------------------------------##
## Enrichment top 10% ##
##----------------------------------------------------------------------------##
msigdb_hs <- msigdbr(species = "Homo sapiens")
selected_terms <- c("JAATINEN_HEMATOPOIETIC_STEM_CELL_UP",
"LIM_MAMMARY_STEM_CELL_UP",
"HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION",
"EPPERT_PROGENITOR",
"HALLMARK_HEME_METABOLISM",
"GSE10325_LUPUS_CD4_TCELL_VS_LUPUS_BCELL_UP",
"GSE22886_NAIVE_CD8_TCELL_VS_NKCELL_UP",
"KEGG_NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY",
"BIOCARTA_NKCELLS_PATHWAY",
"GSE29618_MONOCYTE_VS_PDC_UP",
"GSE29618_MONOCYTE_VS_PDC_DAY7_FLU_VACCINE_UP",
"GSE22886_NAIVE_BCELL_VS_MONOCYTE_UP",
"KEGG_INTESTINAL_IMMUNE_NETWORK_FOR_IGA_PRODUCTION",
"HADDAD_B_LYMPHOCYTE_PROGENITOR",
"LEE_EARLY_T_LYMPHOCYTE_UP",
"GSE22886_NAIVE_TCELL_VS_DC_UP"
)
msigdb_sel <- msigdb_hs %>%
filter(gs_name %in% selected_terms) %>%
mutate(term = gs_name) %>%
mutate(gene = gene_symbol) %>%
select(term, gene)
sign_compare_t10_Msig <- compareCluster(geneClusters = sign_features,
fun = "enricher",
TERM2GENE = msigdb_sel)
dotplot(sign_compare_t10_Msig, showCategory = 30)
Click for Answer