[BC]2 Tutorial
Defining genomic signatures with Non-Negative Matrix Factorization
Carl Herrmann & Andres Quintero
13 September 2021Signature idenfication
After completing the matrix decomposition step and selecting a candidate optimal factorization rank, the matrix H can be visualized to explore the signatures, the signature quality can be assessed using a riverplot visualization, and the association of a signature with a biological or clinical variable can be inferred with a recovery analysis.
H Matrix sample exposure:
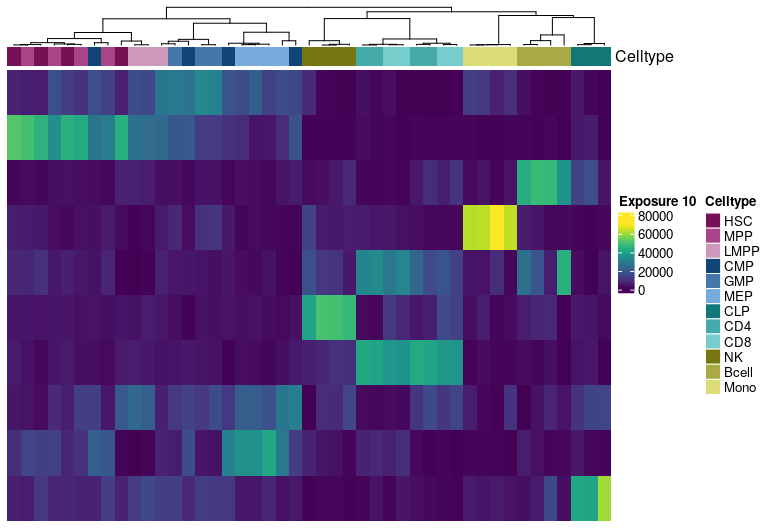
Visualize exposure of the samples to the decomposed signatures as a heatmap, for a selected factorization rank.
##----------------------------------------------------------------------------##
## H matrix heatmap annotation ##
##----------------------------------------------------------------------------##
#Annotation for H matrix heatmap
corces_rna_annot_tmp <- corces_rna_annot %>%
mutate(Celltype = factor(Celltype, levels = c("HSC", "MPP", "LMPP",
"CMP", "GMP", "MEP",
"CLP", "CD4", "CD8",
"NK", "Bcell", "Mono")))
type.colVector <- corces_rna_annot_tmp %>%
select(Celltype, color) %>%
arrange(Celltype) %>%
distinct() %>%
deframe()
type.colVector <- list(Celltype = type.colVector)
# Build Heatmap annotation
heat.anno <- HeatmapAnnotation(df = corces_rna_annot_tmp[,"Celltype",drop=FALSE],
col = type.colVector,
show_annotation_name = TRUE, na_col = "white")
##----------------------------------------------------------------------------##
## Generate H matrix heatmap, W normalized ##
##----------------------------------------------------------------------------##
ki <- 8
tmp.hmatrix <- HMatrix(rna_norm_nmf_exp, k = ki)
Heatmap(tmp.hmatrix,
col = viridis(100),
name = paste0("Exposure ", ki),
clustering_distance_columns = 'pearson',
show_column_dend = TRUE,
heatmap_legend_param =
list(color_bar = "continuous", legend_height=unit(2, "cm")),
top_annotation = heat.anno,
show_column_names = FALSE,
show_row_names = FALSE,
cluster_rows = FALSE)
Click for Answer
H matrix for k= 2
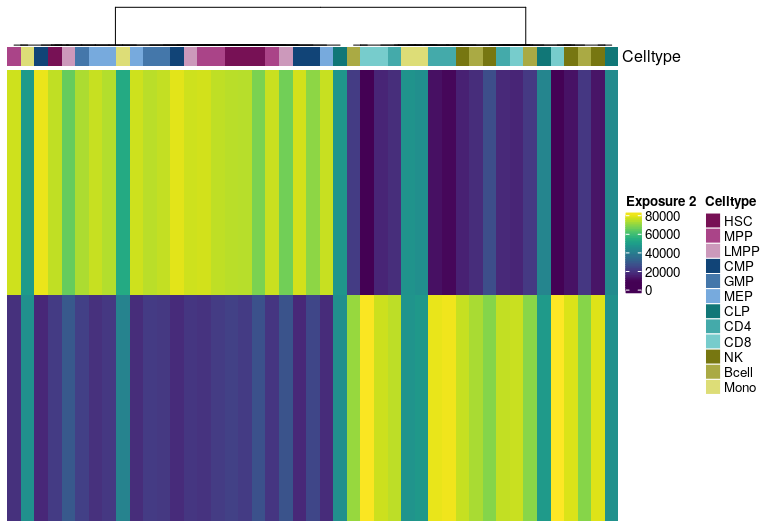
H matrix for k= 3
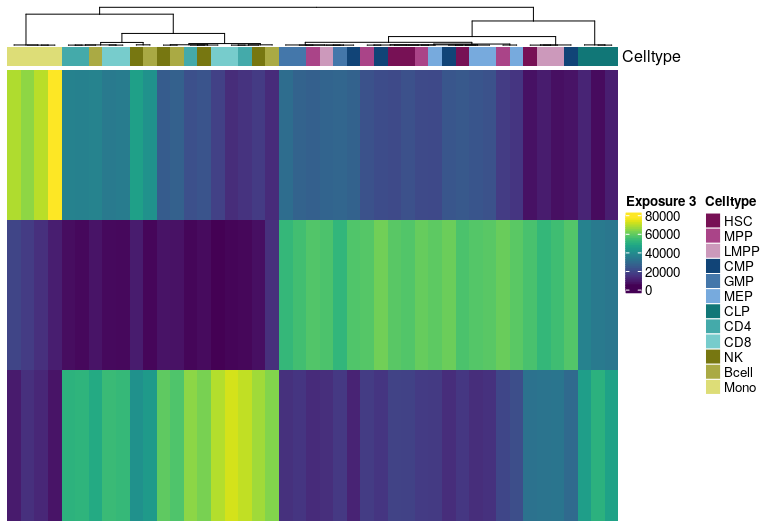
H matrix for k= 4
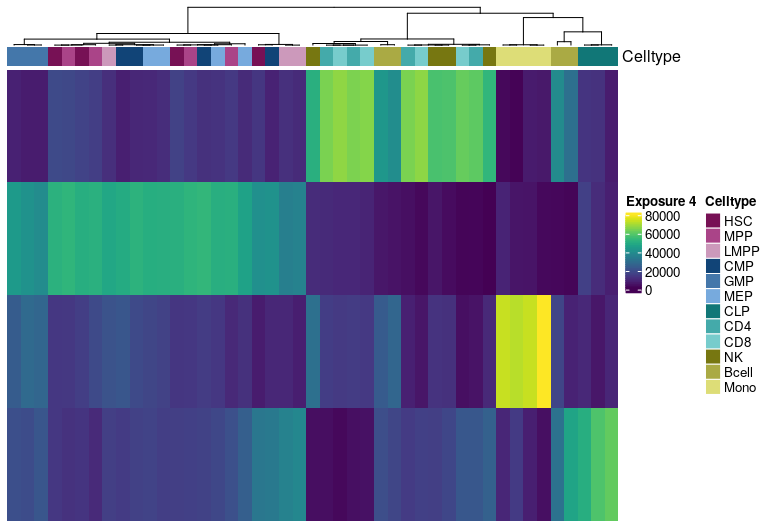
H matrix for k= 5
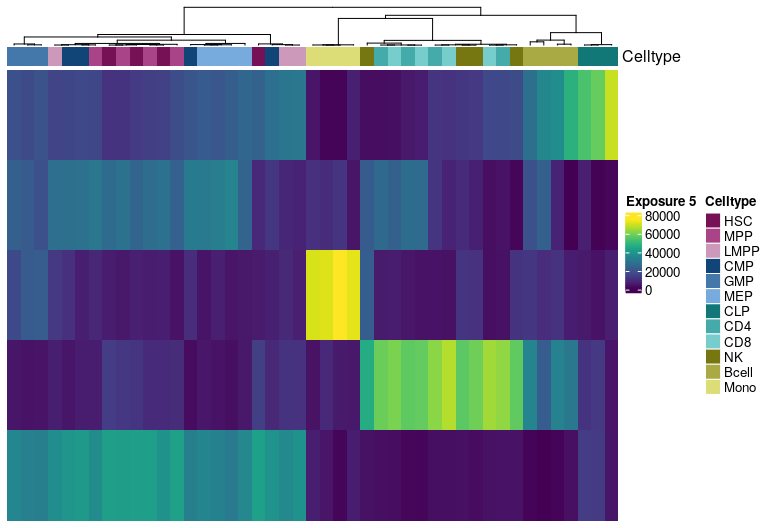
H matrix for k= 6
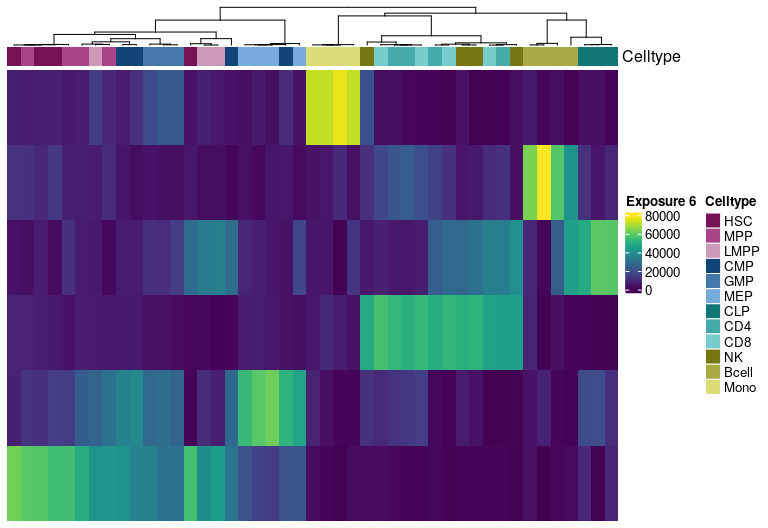
H matrix for k= 7
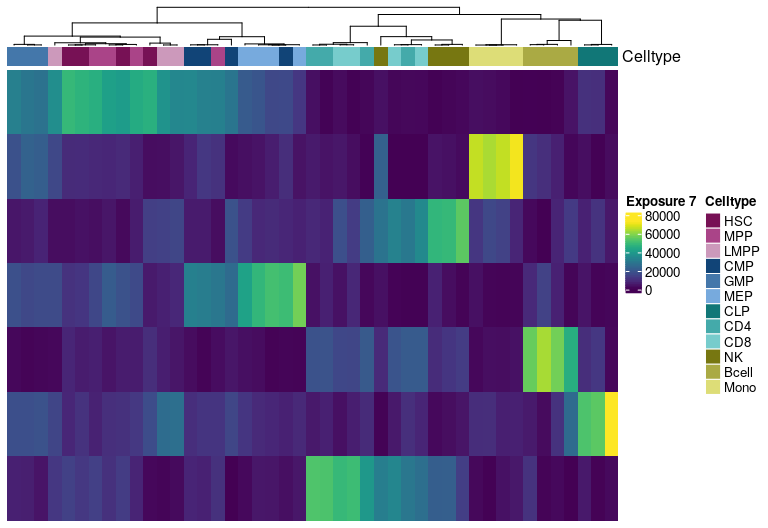
H matrix for k= 8
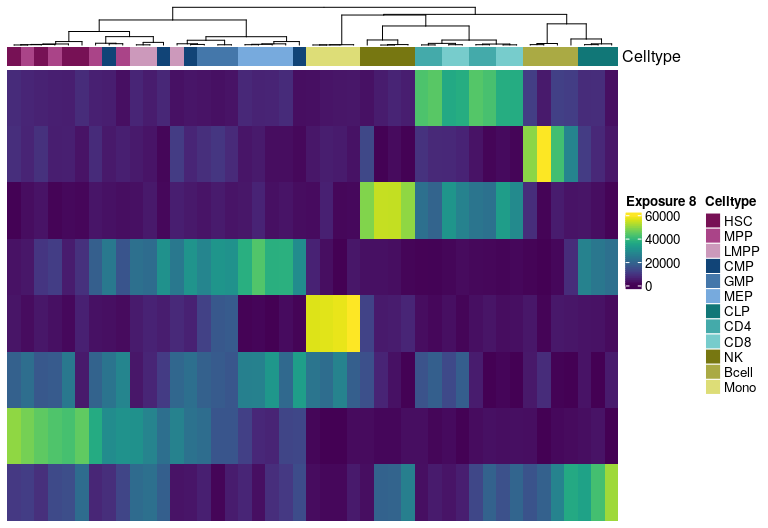
H matrix for k= 9
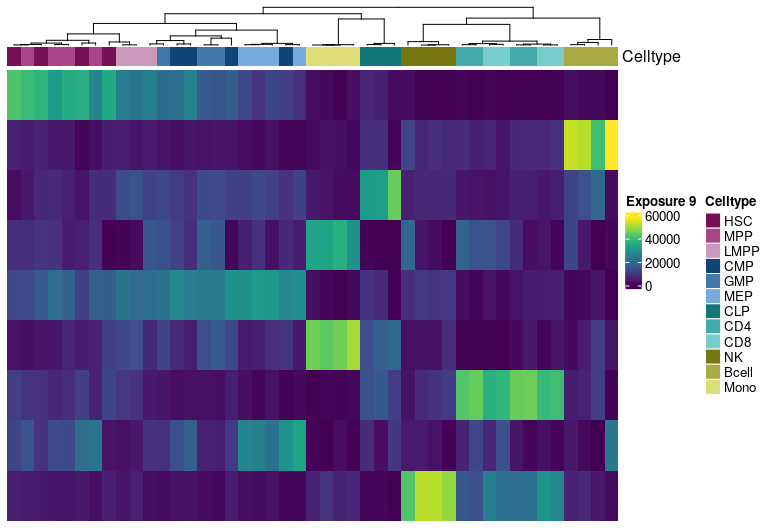
H matrix for k= 10
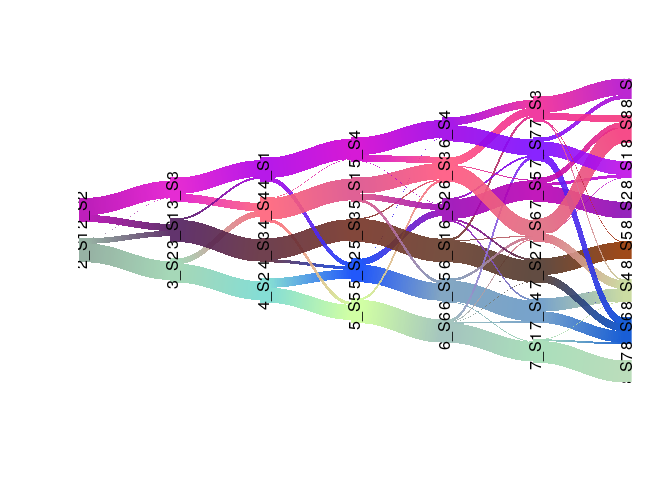
Signature estability - Riverplot visualization
Riverplot representation of the extracted signatures at different factorization ranks. The nodes represent the signatures, the edge strength encodes cosine similarity between signatures linked by the edges.
river <- generateRiverplot(rna_norm_nmf_exp, ranks = 2:8)
plot(river, plot_area=1, yscale=0.6, nodewidth=0.5)
Click for Answer
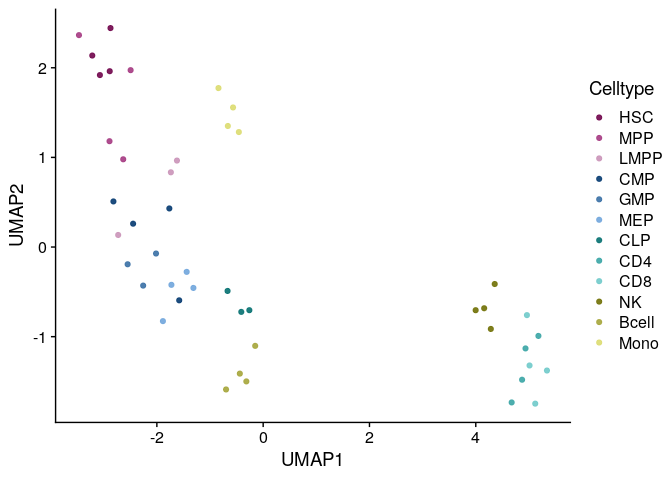
Cluster identification - UMAP
A common practice to identify clusters of samples in single-cell and bulk omics datasets is to perform a dimensionality reduction step with PCA, followed by tSNE or UMAP. The resulting matrix H from the NMF decomposition can be used in the same fashion.
Cluster identification by running UMAP on the matrix H:
##----------------------------------------------------------------------------##
## UMAP H matrix ##
##----------------------------------------------------------------------------##
hmatrix_norm <- HMatrix(rna_norm_nmf_exp, k = 8)
umapView <- umap(t(hmatrix_norm))
umapView_df <- as.data.frame(umapView$layout)
colnames(umapView_df) <- c("UMAP1", "UMAP2")
type_colVector <- corces_rna_annot %>%
dplyr::select(Celltype, color) %>%
arrange(Celltype) %>%
distinct() %>%
deframe()
umapView_df %>%
rownames_to_column("sampleID") %>%
left_join(corces_rna_annot, by = "sampleID") %>%
mutate(Celltype = factor(Celltype, levels = c("HSC", "MPP", "LMPP",
"CMP", "GMP", "MEP",
"CLP", "CD4", "CD8",
"NK", "Bcell", "Mono"))) %>%
ggplot(aes(x=UMAP1, y=UMAP2, color = Celltype)) +
geom_point(size = 1.5, alpha = 0.95) +
scale_color_manual(values = type_colVector) +
theme_cowplot()
Click for Answer
Association of signatures to biological variables
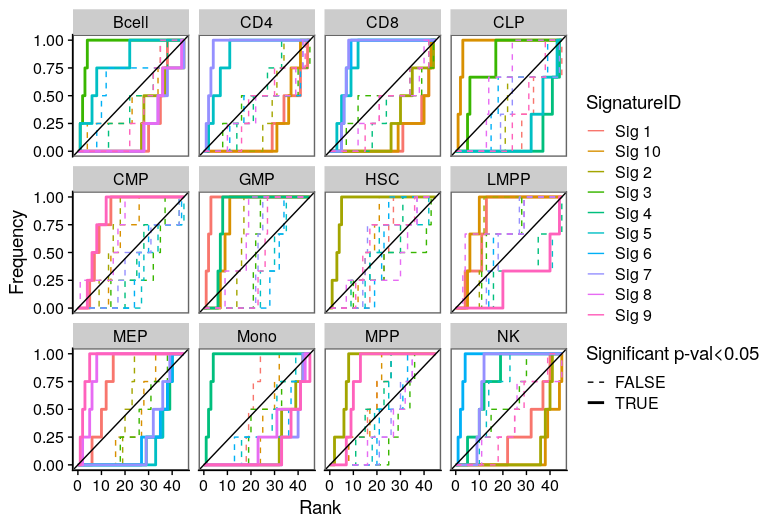
One of the most important steps in the identification of molecular signatures, is to find the association of a candidate signature with know biological or clinical variables. In ButchR, it is possible to generate a recovery plot which intuitively shows the degree of association between all the signatures for one factorization rank and one known biological variable.
Make a recovery plot showing the association of the NMF signatures (for a selected factorization rank) to the "Celltype" variable:
##----------------------------------------------------------------------------##
## Recovery plots ##
##----------------------------------------------------------------------------##
ki <- 8
tmp.hmatrix <- HMatrix(rna_norm_nmf_exp, k = ki)
ButchR::recovery_plot(tmp.hmatrix, corces_rna_annot$Celltype)
Click for Answer
Recovery plots for k= 2
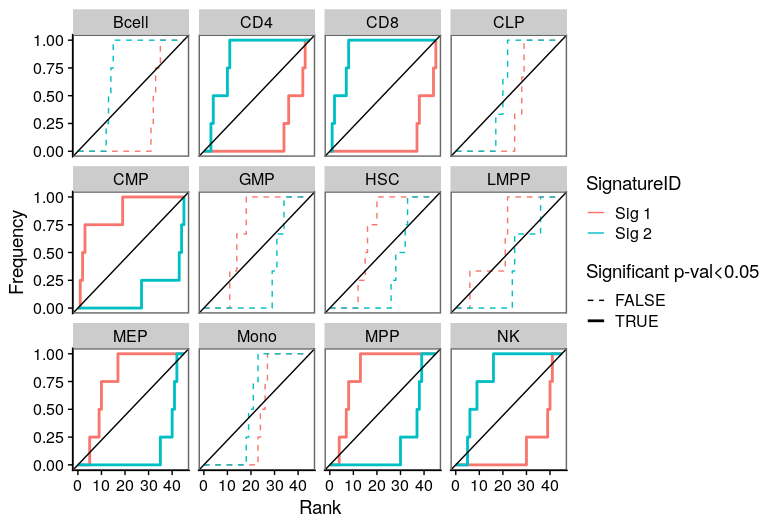
Recovery plots for k= 3
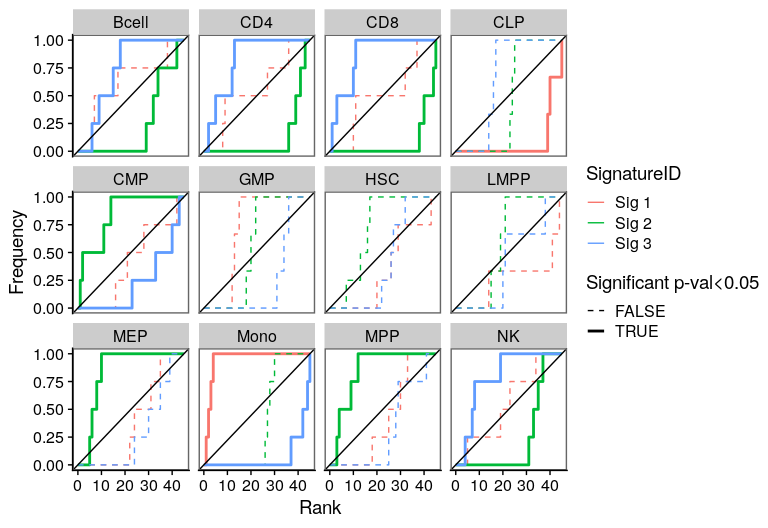
Recovery plots for k= 4
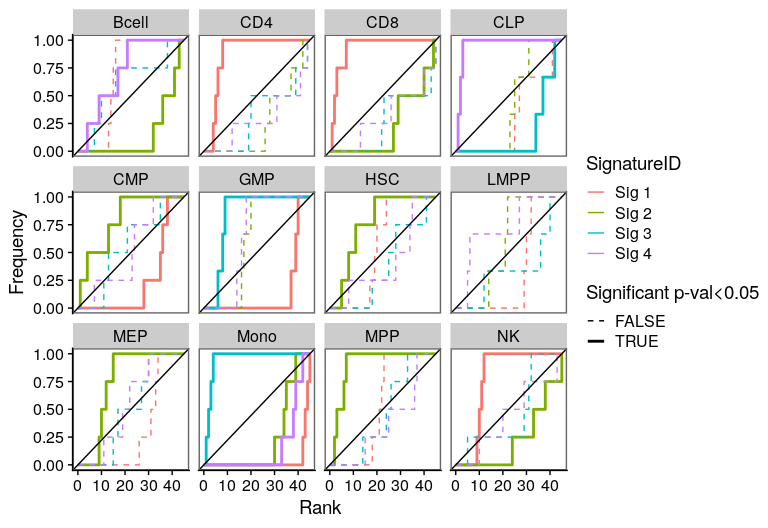
Recovery plots for k= 5
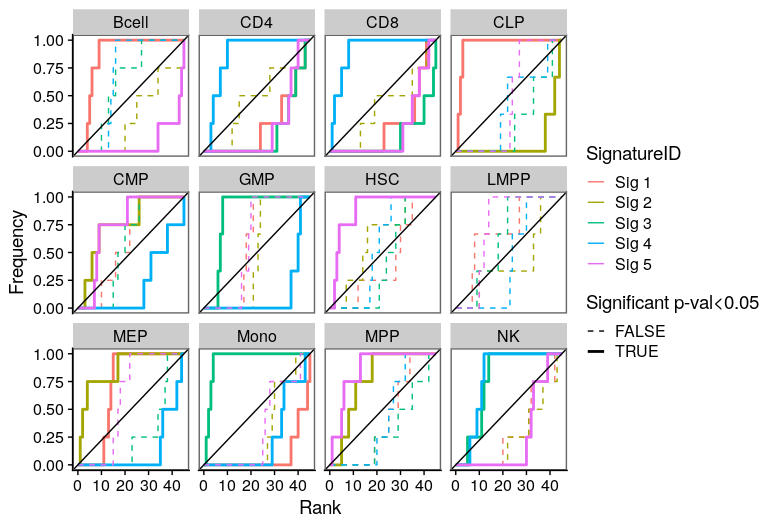
Recovery plots for k= 6
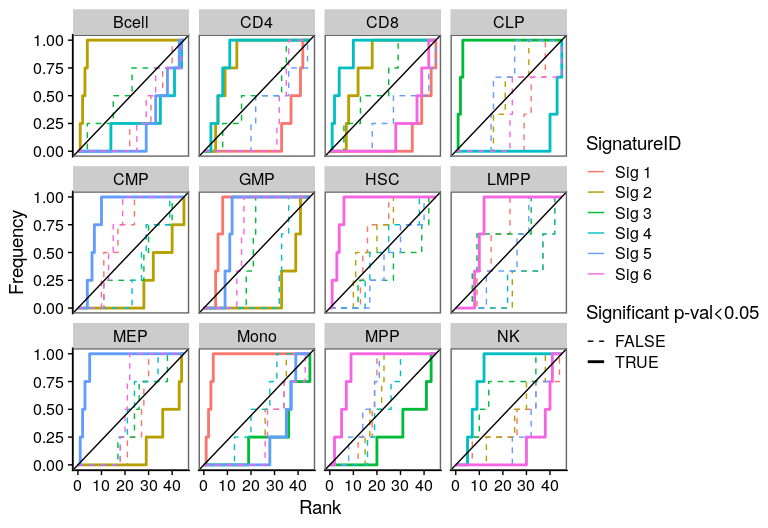
Recovery plots for k= 7
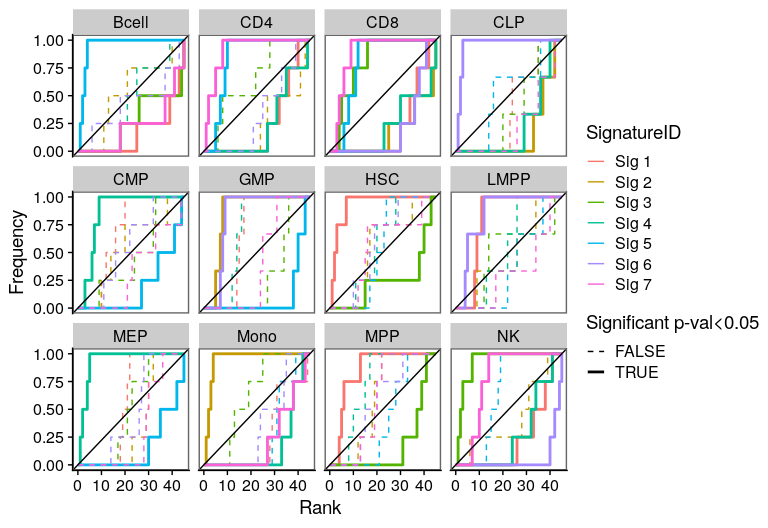
Recovery plots for k= 8
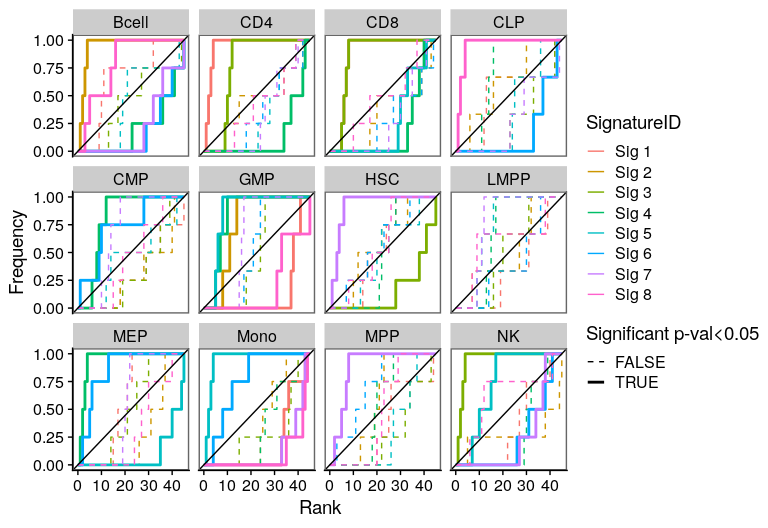
Recovery plots for k= 9
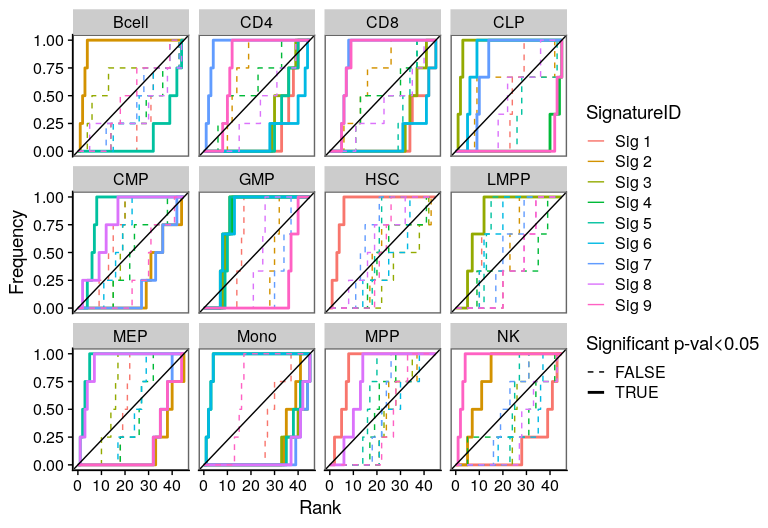
Recovery plots for k= 10